भाग 1: एक डेमो पाइपलाइन चलाएं¶
AI-सहायता प्राप्त अनुवाद - अधिक जानें और सुधार सुझाएं
Hello nf-core प्रशिक्षण पाठ्यक्रम के इस पहले भाग में, हम तुम्हें दिखाएंगे कि एक nf-core pipeline कैसे खोजें और आज़माएं, अपनी ज़रूरतों के अनुसार इसके execution को कैसे configure और customize करें, और यह समझें कि input validation सामान्य त्रुटियों से कैसे बचाता है।
हम nf-core/demo नामक एक pipeline का उपयोग करने जा रहे हैं जिसे nf-core प्रोजेक्ट अपने pipelines की सूची के हिस्से के रूप में demonstration और प्रशिक्षण उद्देश्यों के लिए बनाए रखता है।
सुनिश्चित करें कि तुम्हारी वर्किंग डायरेक्टरी hello-nf-core/ पर सेट है जैसा कि Getting started पेज पर निर्देश दिया गया है।
1. nf-core/demo pipeline खोजें और प्राप्त करें¶
आइए nf-co.re पर प्रोजेक्ट वेबसाइट पर nf-core/demo pipeline को खोजने से शुरू करें, जो सभी जानकारी को केंद्रीकृत करती है जैसे: सामान्य दस्तावेज़ीकरण और सहायता लेख, प्रत्येक pipeline के लिए दस्तावेज़ीकरण, ब्लॉग पोस्ट, इवेंट की घोषणाएं इत्यादि।
1.1. वेबसाइट पर pipeline खोजें¶
अपने वेब ब्राउज़र में, https://nf-co.re/pipelines/ पर जाओ और सर्च बार में demo टाइप करो।
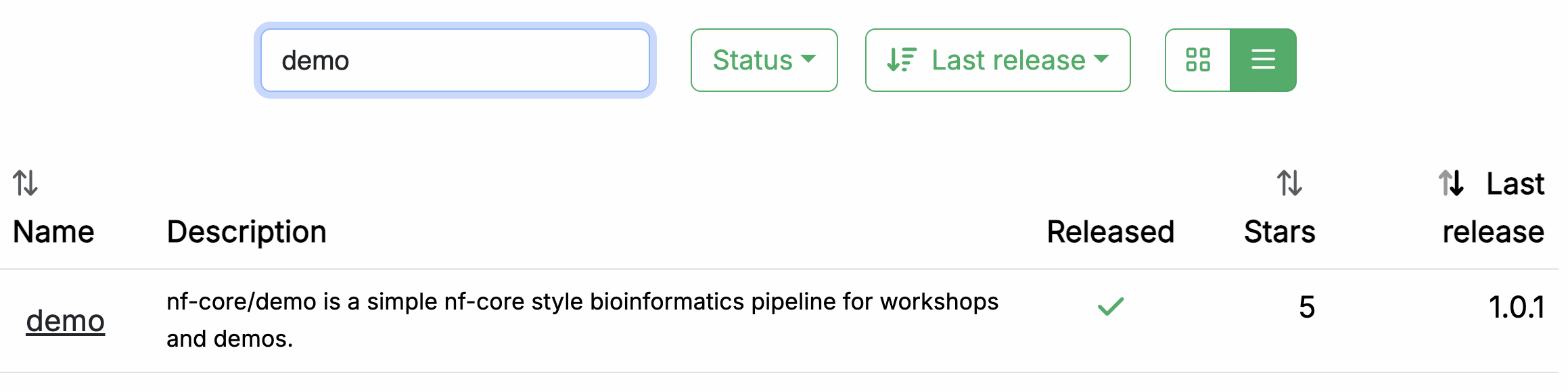
Pipeline दस्तावेज़ीकरण पेज तक पहुँचने के लिए pipeline के नाम, demo, पर क्लिक करो।
प्रत्येक रिलीज़ की गई pipeline का एक समर्पित पेज होता है जिसमें निम्नलिखित दस्तावेज़ीकरण अनुभाग शामिल होते हैं:
- Introduction: Pipeline का परिचय और अवलोकन
- Usage: Pipeline को execute करने के तरीके का विवरण
- Parameters: विवरण के साथ समूहीकृत pipeline पैरामीटर
- Output: अपेक्षित आउटपुट फ़ाइलों का विवरण और उदाहरण
- Results: पूर्ण test dataset से उत्पन्न उदाहरण आउटपुट फ़ाइलें
- Releases & Statistics: Pipeline संस्करण इतिहास और आंकड़े
जब भी तुम एक नई pipeline को अपनाने पर विचार कर रहे हो, तो पहले pipeline दस्तावेज़ीकरण को ध्यान से पढ़ो ताकि यह समझ सको कि यह क्या करती है और इसे चलाने का प्रयास करने से पहले इसे कैसे कॉन्फ़िगर किया जाना चाहिए।
अभी देखो और पता लगाओ:
- Pipeline कौन से टूल चलाएगी (टैब देखें:
Introduction) - Pipeline कौन से इनपुट और पैरामीटर स्वीकार करती है या आवश्यक है (टैब देखें:
Parameters) - Pipeline द्वारा उत्पादित आउटपुट क्या हैं (टैब देखें:
Output)
1.1.1. Pipeline अवलोकन¶
Introduction टैब pipeline का एक अवलोकन प्रदान करता है, जिसमें एक दृश्य प्रतिनिधित्व (जिसे subway map कहा जाता है) और pipeline के हिस्से के रूप में चलाए जाने वाले टूल की एक सूची शामिल है।

- Read QC (FASTQC)
- Adapter and quality trimming (SEQTK_TRIM)
- Present QC for raw reads (MULTIQC)
1.1.2. उदाहरण कमांड लाइन¶
दस्तावेज़ीकरण एक उदाहरण इनपुट फ़ाइल (जिसकी आगे चर्चा की जाएगी) और एक उदाहरण कमांड लाइन भी प्रदान करता है।
nextflow run nf-core/demo \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>
तुम देखोगे कि उदाहरण कमांड एक workflow फ़ाइल निर्दिष्ट नहीं करती है, केवल pipeline रिपॉज़िटरी का संदर्भ, nf-core/demo।
इस तरह से शुरू करने पर, Nextflow मान लेगा कि कोड एक निश्चित तरीके से व्यवस्थित है। आइए कोड प्राप्त करें ताकि हम इस संरचना की जांच कर सकें।
1.2. Pipeline कोड प्राप्त करें¶
एक बार जब हम यह निर्धारित कर लेते हैं कि pipeline हमारे उद्देश्यों के लिए उपयुक्त प्रतीत होती है, तो आइए इसे आज़माएं। सौभाग्य से Nextflow सही ढंग से प्रारूपित रिपॉज़िटरी से pipelines को मैन्युअल रूप से कुछ भी डाउनलोड किए बिना प्राप्त करना आसान बनाता है।
1.2.1. nextflow pull का उपयोग करें¶
आइए टर्मिनल पर वापस जाएं और निम्नलिखित चलाएं:
कमांड आउटपुट
Nextflow pipeline कोड का एक pull करता है, मतलब यह पूरी रिपॉज़िटरी को तुम्हारी लोकल ड्राइव पर डाउनलोड करता है।
स्पष्ट करने के लिए, तुम GitHub में उचित रूप से सेट की गई किसी भी Nextflow pipeline के साथ ऐसा कर सकते हो, न केवल nf-core pipelines के साथ। हालाँकि nf-core Nextflow pipelines का सबसे बड़ा ओपन-सोर्स संग्रह है।
1.2.2. nextflow list का उपयोग करें¶
तुम Nextflow से इस तरह से प्राप्त की गई pipelines की सूची दे सकते हो:
तुम कुछ और pipelines pull करके देख सकते हो कि जब एक से अधिक हों तो वे कैसे सूचीबद्ध होती हैं।
1.2.3. $NXF_HOME/assets/ में अपनी pipelines खोजें¶
तुम देखोगे कि फ़ाइलें तुम्हारी वर्तमान कार्य डायरेक्टरी में नहीं हैं।
डिफ़ॉल्ट रूप से, Nextflow उन्हें $NXF_HOME/assets में सहेजता है।
नोट
यदि तुम हमारे प्रशिक्षण वातावरण का उपयोग नहीं कर रहे हो तो तुम्हारे सिस्टम पर पूर्ण पथ भिन्न हो सकता है।
Nextflow डाउनलोड किए गए स्रोत कोड को जानबूझकर 'बाहर' रखता है इस सिद्धांत पर कि इन pipelines का उपयोग उस कोड की तुलना में अधिक लाइब्रेरी की तरह किया जाना चाहिए जिसके साथ तुम सीधे इंटरैक्ट करोगे।
1.2.4. स्रोत कोड तक आसानी से पहुँचने के लिए एक symlink बनाएं¶
हम कोड को विस्तार से नहीं देखेंगे, लेकिन समग्र संगठन कैसा दिखता है इसका एक त्वरित अवलोकन लेते हैं।
Pipeline स्रोत कोड को ब्राउज़ करना आसान बनाने के लिए, assets डायरेक्टरी के लिए एक symbolic link बनाओ:
यह एक शॉर्टकट बनाता है ताकि तुम tree -L 2 pipelines से कोड explore कर सको या फ़ाइलें सीधे खोल सको।
1.2.5. कोड संगठन का अवलोकन¶
तुम या तो tree का उपयोग कर सकते हो या nf-core/demo डायरेक्टरी को खोजने और खोलने के लिए फ़ाइल एक्सप्लोरर का उपयोग कर सकते हो।
डायरेक्टरी सामग्री
pipelines/nf-core/demo
├── assets
├── CHANGELOG.md
├── CITATIONS.md
├── CODE_OF_CONDUCT.md
├── conf
├── docs
├── LICENSE
├── main.nf
├── modules
├── modules.json
├── nextflow.config
├── nextflow_schema.json
├── nf-test.config
├── README.md
├── ro-crate-metadata.json
├── subworkflows
├── tests
├── tower.yml
└── workflows
जैसा कि तुम देख सकते हो, वहाँ बहुत कुछ चल रहा है, जिसके बारे में अधिकांश की चिंता करने की ज़रूरत नहीं है।
संक्षेप में, ध्यान दें कि शीर्ष स्तर पर, तुम सारांश जानकारी के साथ एक README फ़ाइल पा सकते हो, साथ ही साथ सहायक फ़ाइलें जो प्रोजेक्ट की जानकारी जैसे लाइसेंसिंग, योगदान दिशानिर्देश, उद्धरण और आचार संहिता को सारांशित करती हैं।
विस्तृत pipeline दस्तावेज़ीकरण docs डायरेक्टरी में स्थित है।
यह सभी सामग्री nf-core वेबसाइट पर वेब पेज उत्पन्न करने के लिए प्रोग्रामेटिक रूप से उपयोग की जाती है, इसलिए वे हमेशा कोड के साथ अद्यतित रहते हैं।
बाकी के लिए, हम कोड फ़ाइलों के तीन कार्यात्मक समूहों को अलग कर सकते हैं:
- Pipeline कोड घटक (
main.nf,workflows,subworkflows,modules) - Pipeline कॉन्फ़िगरेशन
- Pipeline पैरामीटर / इनपुट और सत्यापन
हम इस भाग में pipeline कोड घटकों पर नहीं जाएंगे, लेकिन हम कॉन्फ़िगरेशन और सत्यापन के उन तत्वों को छुएंगे जो nf-core pipelines के अंतिम उपयोगकर्ता के रूप में तुम्हारे लिए प्रासंगिक होने की संभावना है।
सुझाव
तुम किसी भी nf-core pipeline का स्रोत कोड GitHub पर भी ब्राउज़ कर सकते हो, जैसे github.com/nf-core/demo। हर nf-core pipeline एक ही डायरेक्टरी layout का पालन करती है, इसलिए एक बार जब तुम संरचना जान लो, तो किसी भी pipeline के लिए configuration फ़ाइलें, modules और workflows उसी तरह खोज सकते हो।
लेकिन अभी के लिए, pipeline चलाने की ओर बढ़ते हैं!
सारांश¶
अब तुम जानते हो कि nf-core वेबसाइट के माध्यम से एक pipeline कैसे खोजें और स्रोत कोड की एक स्थानीय प्रति कैसे प्राप्त करें।
आगे क्या है?¶
सीखो कि न्यूनतम प्रयास के साथ एक nf-core pipeline कैसे आज़माएं।
2. अपने test profile के साथ pipeline आज़माएं¶
सुविधाजनक रूप से, प्रत्येक nf-core pipeline एक test profile के साथ आती है। यह nf-core/test-datasets रिपॉज़िटरी में होस्ट किए गए एक छोटे test dataset का उपयोग करके चलने के लिए pipeline के लिए कॉन्फ़िगरेशन सेटिंग्स का एक न्यूनतम सेट है। यह छोटे पैमाने पर एक pipeline को जल्दी से आज़माने का एक शानदार तरीका है।
नोट
Nextflow का configuration profile सिस्टम तुम्हें विभिन्न कंटेनर इंजन या execution वातावरण के बीच आसानी से स्विच करने की अनुमति देता है। अधिक विवरण के लिए, Hello Nextflow भाग 6: Configuration देखें।
2.1. test profile की जांच करें¶
Pipeline के test profile को चलाने से पहले यह जांचना अच्छा अभ्यास है कि यह क्या निर्दिष्ट करता है।
nf-core/demo के लिए test profile कॉन्फ़िगरेशन फ़ाइल conf/test.config में रहता है।
तुम इसे pipeline स्रोत के अंदर locally खोज सकते हो जो nextflow pull ने डाउनलोड किया:
उस फ़ाइल की सामग्री यहाँ दी गई है:
तुम तुरंत देखोगे कि शीर्ष पर कमेंट ब्लॉक में एक उपयोग उदाहरण शामिल है जो दिखाता है कि इस test profile के साथ pipeline को कैसे चलाना है।
| conf/test.config | |
|---|---|
हमें केवल वही प्रदान करने की आवश्यकता है जो उदाहरण कमांड में कैरेट के बीच दिखाया गया है: <docker/singularity> और <OUTDIR>।
याद दिलाने के लिए, <docker/singularity> कंटेनर सिस्टम की पसंद को संदर्भित करता है। सभी nf-core pipelines को reproducibility सुनिश्चित करने और सॉफ़्टवेयर इंस्टॉलेशन समस्याओं को खत्म करने के लिए कंटेनर (Docker, Singularity, आदि) के साथ उपयोग करने योग्य होने के लिए डिज़ाइन किया गया है।
इसलिए हमें यह निर्दिष्ट करना होगा कि हम pipeline का परीक्षण करने के लिए Docker या Singularity का उपयोग करना चाहते हैं या नहीं।
--outdir <OUTDIR> भाग उस डायरेक्टरी को संदर्भित करता है जहाँ Nextflow pipeline के आउटपुट लिखेगा।
हमें इसके लिए एक नाम प्रदान करने की आवश्यकता है, जिसे हम बस बना सकते हैं।
यदि यह पहले से मौजूद नहीं है, तो Nextflow इसे runtime पर हमारे लिए बना देगा।
कमेंट ब्लॉक के बाद के अनुभाग की ओर बढ़ते हुए, test profile हमें दिखाता है कि परीक्षण के लिए क्या पूर्व-कॉन्फ़िगर किया गया है: सबसे विशेष रूप से, input पैरामीटर पहले से ही एक test dataset की ओर इशारा करने के लिए सेट है, इसलिए हमें अपना डेटा प्रदान करने की आवश्यकता नहीं है।
यदि तुम पूर्व-कॉन्फ़िगर किए गए इनपुट के लिंक का अनुसरण करते हो, तो तुम देखोगे कि यह एक csv फ़ाइल है जिसमें कई प्रायोगिक नमूनों के लिए नमूना पहचानकर्ता और फ़ाइल पथ हैं।
sample,fastq_1,fastq_2
SAMPLE1_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R2.fastq.gz
SAMPLE2_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R2.fastq.gz
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,
इसे samplesheet कहा जाता है, और यह nf-core pipelines के लिए इनपुट का सबसे आम रूप है।
नोट
यदि तुम डेटा प्रारूप और प्रकारों से परिचित नहीं हो तो चिंता मत करो, यह आगे के लिए महत्वपूर्ण नहीं है।
तो यह पुष्टि करता है कि हमारे पास pipeline को आज़माने के लिए आवश्यक सब कुछ है।
2.2. Pipeline चलाएं¶
आइए कंटेनर सिस्टम के लिए Docker का उपयोग करने और आउटपुट डायरेक्टरी के रूप में demo-results का निर्णय लें, और हम test कमांड चलाने के लिए तैयार हैं:
कमांड आउटपुट
N E X T F L O W ~ version 25.10.4
Launching `https://github.com/nf-core/demo` [magical_pauling] DSL2 - revision: 45904cb9d1 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.1.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2025-11-21_04-57-41
Core Nextflow options
revision : master
runName : magical_pauling
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/nf-core/demo
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/nf-core/demo/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (7)
[ff/a6976b] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) | 3 of 3 ✔
[39/731ab7] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) | 3 of 3 ✔
[7c/78d96e] NFCORE_DEMO:DEMO:MULTIQC | 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
यदि तुम्हारा आउटपुट उससे मेल खाता है, तो बधाई हो! तुमने अभी अपनी पहली nf-core pipeline चलाई है।
तुम देखोगे कि जब तुम एक बुनियादी Nextflow pipeline चलाते हो तो कंसोल आउटपुट बहुत अधिक है। एक हेडर है जिसमें pipeline के संस्करण, इनपुट और आउटपुट, और कॉन्फ़िगरेशन के कुछ तत्वों का सारांश शामिल है।
नोट
तुम्हारा आउटपुट अलग-अलग timestamps, execution नाम और फ़ाइल पथ दिखाएगा, लेकिन समग्र संरचना और process execution समान होनी चाहिए।
आउटपुट के शीर्ष के पास इस लाइन पर ध्यान दो:
यह तुम्हें बताता है कि pipeline का कौन सा revision उपयोग किया गया था।
क्योंकि हमने कोई संस्करण निर्दिष्ट नहीं किया, Nextflow ने master पर नवीनतम commit का उपयोग किया।
Reproducible runs के लिए, तुम्हें -r flag का उपयोग करके एक विशिष्ट release pin करनी चाहिए:
यह सुनिश्चित करता है कि नए commits या releases की परवाह किए बिना हर बार एक ही pipeline कोड का उपयोग किया जाए।
इस प्रशिक्षण के लिए हम सरलता के लिए -r को छोड़ देते हैं, लेकिन production में तुम्हें हमेशा इसे निर्दिष्ट करना चाहिए।
Execution आउटपुट की ओर बढ़ते हुए, आइए उन लाइनों पर एक नज़र डालें जो हमें बताती हैं कि कौन से processes चलाए गए थे:
executor > local (7)
[ff/a6976b] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) | 3 of 3 ✔
[39/731ab7] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) | 3 of 3 ✔
[7c/78d96e] NFCORE_DEMO:DEMO:MULTIQC | 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
यह हमें बताता है कि तीन processes चलाई गईं, जो nf-core वेबसाइट पर pipeline दस्तावेज़ीकरण पेज में दिखाए गए तीन टूल से संबंधित हैं: FASTQC, SEQTK_TRIM और MULTIQC।
पूर्ण process नाम जैसा कि यहाँ दिखाया गया है, जैसे NFCORE_DEMO:DEMO:MULTIQC, परिचयात्मक Hello Nextflow सामग्री में तुमने जो देखा होगा उससे लंबे हैं।
इनमें उनके parent workflows के नाम शामिल हैं और pipeline कोड की modularity को दर्शाते हैं।
हम इसके बारे में इस पाठ्यक्रम के भाग 2 में अधिक विस्तार से बात करेंगे।
2.3. Pipeline के आउटपुट की जांच करें¶
अंत में, आइए pipeline द्वारा उत्पादित demo-results डायरेक्टरी पर एक नज़र डालें।
डायरेक्टरी सामग्री
demo-results
├── fastqc
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── fq
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── multiqc
│ ├── multiqc_data
│ ├── multiqc_plots
│ └── multiqc_report.html
└── pipeline_info
├── execution_report_2025-11-21_04-57-41.html
├── execution_timeline_2025-11-21_04-57-41.html
├── execution_trace_2025-11-21_04-57-41.txt
├── nf_core_demo_software_mqc_versions.yml
├── params_2025-11-21_04-57-46.json
└── pipeline_dag_2025-11-21_04-57-41.html
यह बहुत अधिक लग सकता है।
nf-core/demo pipeline के आउटपुट के बारे में अधिक जानने के लिए, इसका दस्तावेज़ीकरण पेज देखें।
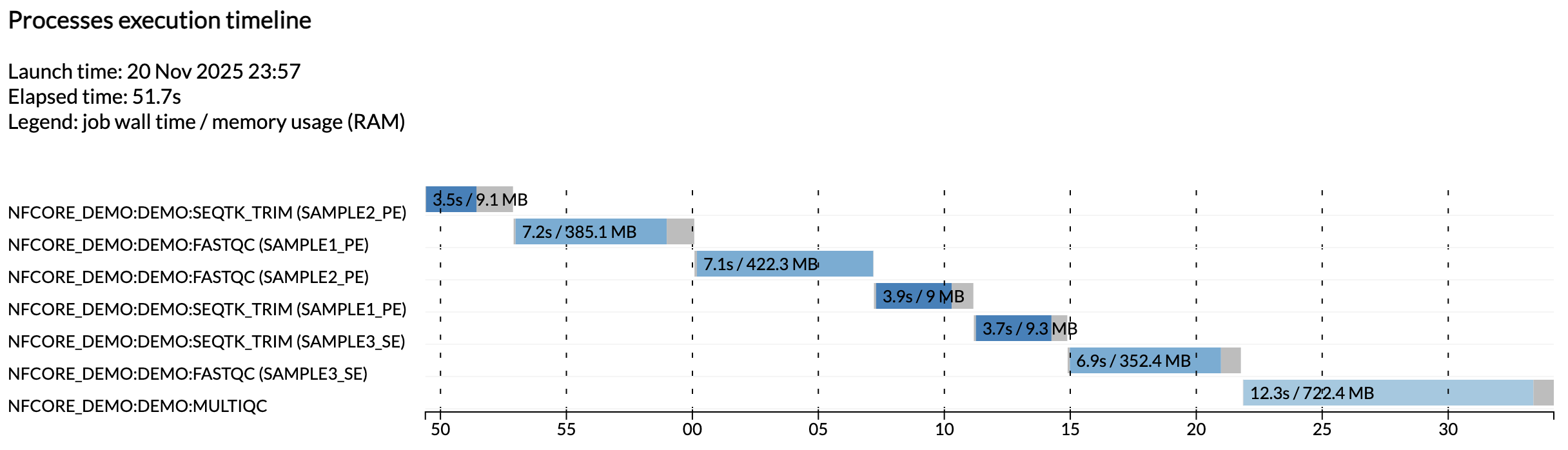
इस स्तर पर, यह देखना महत्वपूर्ण है कि परिणाम module द्वारा व्यवस्थित हैं, और इसके अतिरिक्त pipeline_info नामक एक डायरेक्टरी है जिसमें pipeline execution के बारे में विभिन्न timestamped रिपोर्ट हैं।
उदाहरण के लिए, execution_timeline_* फ़ाइल तुम्हें दिखाती है कि कौन से processes चलाए गए, किस क्रम में और उन्हें चलाने में कितना समय लगा:
नोट
यहाँ कार्य समानांतर में नहीं चलाए गए क्योंकि हम Github Codespaces में एक minimalist मशीन पर चल रहे हैं। इन्हें समानांतर में चलते हुए देखने के लिए, अपने codespace के CPU allocation और test configuration में resource limits को बढ़ाने का प्रयास करो।
ये रिपोर्ट सभी nf-core pipelines के लिए स्वचालित रूप से उत्पन्न होती हैं।
सारांश¶
तुम जानते हो कि अपने अंतर्निहित test profile का उपयोग करके एक nf-core pipeline कैसे चलाएं और इसके आउटपुट कहाँ खोजें।
आगे क्या है?¶
सीखो कि pipeline के execution को customize करने के लिए इसे कैसे configure करें।
3. Pipeline execution को configure करें¶
जैसा कि Hello Config में समझाया गया है, हम चाहते हैं कि pipeline कोड को बदले बिना यह बदल सकें कि हमारी pipeline किस डेटा पर चलेगी और कैसे चलेगी। इसके लिए, Nextflow pipeline configuration को नियंत्रित करने के कई तरीके प्रदान करता है, जो थोड़ा overwhelming हो सकता है।
nf-core प्रोजेक्ट configuration तत्वों को व्यवस्थित करने के लिए conventions निर्दिष्ट करता है, शीर्ष स्तर पर दो प्रकार के configuration को अलग करता है: pipeline parameters और सख्त अर्थ में configuration।
- Pipeline parameters (
paramsसिस्टम के माध्यम से सेट) में आमतौर पर इनपुट फ़ाइलें, टूल व्यवहार flags और विश्लेषण पैरामीटर जैसी चीज़ें शामिल होती हैं। - सख्त अर्थ में Configuration pipeline के चलने के तरीके की logistics को संदर्भित करता है, यानी executor, compute resource allocations आदि।
आइए pipeline parameters से शुरू करें, फिर हम सख्त अर्थ में configuration को देखेंगे।
3.1. Pipeline parameters¶
सभी nf-core pipelines के लिए, तुम --help flag का उपयोग करके सीधे कमांड लाइन से pipeline parameters की पूरी सूची प्राप्त कर सकते हो, जो स्वयं एक pipeline parameter है।
3.1.1. --help के साथ parameters की सूची प्राप्त करें¶
Demo pipeline के लिए help कमांड चलाओ:
कमांड आउटपुट
N E X T F L O W ~ version 25.10.4
Launching `https://github.com/nf-core/demo` [run_name] DSL2 - revision: 45904cb9d1 [master]
----------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.1.0
----------------------------------------------------
Typical pipeline command:
nextflow run nf-core/demo -profile <docker/singularity/.../institute> --input samplesheet.csv --outdir <OUTDIR>
Input/output options
--input [string] Path to a metadata file containing information about the samples in the experiment.
--outdir [string] The output directory where the results will be saved. You have to use absolute paths to storage on Cloud infrastructure.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header, used for filename if not otherwise specified.
Reference genome options
--genome [string] Name of iGenomes reference.
--fasta [string] Path to FASTA genome file.
Process skipping options
--skip_trim [boolean] Skip trimming fastq files with seqtk
Generic options
--multiqc_methods_description [string] Custom MultiQC yaml file containing HTML including a methods description.
--help [boolean, string] Display the help message.
--help_full [boolean] Display the full detailed help message.
--show_hidden [boolean] Display hidden parameters in the help message (only works when --help or --help_full are provided).
!! Hiding 20 param(s), use the `--show_hidden` parameter to show them !!
----------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
जैसा कि तुम देख सकते हो, आउटपुट parameters को श्रेणियों में समूहित करता है (Input/output options, Reference genome options, आदि) प्रत्येक के लिए प्रकार और विवरण के साथ।
यह वर्गीकरण एक schema फ़ाइल द्वारा निर्धारित किया जाता है, जिसे आगे नीचे कवर किया गया है।
Plain Nextflow pipelines में, --help केवल तभी काम करता है जब developer ने इसे मैन्युअल रूप से implement किया हो।
सुझाव
डिफ़ॉल्ट रूप से छिपे हुए अतिरिक्त parameters देखने के लिए --help --show_hidden का उपयोग करो, जैसे --publish_dir_mode या --monochrome_logs।
3.1.2. Parameter values सेट करें¶
जैसा कि Hello Config में कवर किया गया है, तुम कमांड लाइन पर --param_name के साथ parameter values सेट कर सकते हो या parameters का एक सेट YAML फ़ाइल में एकत्र कर सकते हो और -params-file के साथ पास कर सकते हो।
दोनों तरीके nf-core pipelines के साथ उसी तरह काम करते हैं।
उदाहरण के लिए, trimming चरण को skip करने के लिए:
कमांड आउटपुट
SEQTK_TRIM process अब आउटपुट में नहीं दिखती।
जानकारी
हालांकि तकनीकी रूप से -c के साथ पास की गई custom configuration फ़ाइल में pipeline parameters सेट करना संभव है, यह pipeline के अपने nextflow.config में पहले से सेट किए गए defaults को override नहीं कर सकता, Nextflow के configuration precedence rules के आधार पर।
कमांड लाइन पर --param_name या -params-file का उपयोग करना अधिक विश्वसनीय है, क्योंकि ये हमेशा प्राथमिकता लेते हैं।
एक सामान्य नियम के रूप में: यदि यह --help आउटपुट में दिखता है, तो इसे config फ़ाइल के बजाय कमांड लाइन या params फ़ाइल के माध्यम से सेट करो।
3.1.3. Parameter validation¶
मज़ेदार तथ्य: --help कमांड सभी nf-core pipelines के लिए काम करता है क्योंकि nf-core प्रोजेक्ट developers को सभी pipeline parameters को एक JSON schema फ़ाइल (nextflow_schema.json) में औपचारिक रूप से परिभाषित करने की आवश्यकता होती है।
यह schema प्रत्येक parameter का प्रकार, विवरण, डिफ़ॉल्ट मान और समूहीकरण रिकॉर्ड करता है।
--help आउटपुट को शक्ति देने के अलावा, schema फ़ाइल launch के समय स्वचालित validation को भी सक्षम करती है।
इसका मतलब है कि Nextflow जांच कर सकता है कि तुम्हारे द्वारा पास किया गया प्रत्येक parameter मौजूद है और उसे एक उचित मान दिया गया है (उचित प्रकार का, अनुमत मानों की सीमा के भीतर आदि)।
हम इसे भाग 5: Input Validation में अधिक विस्तार से कवर करते हैं, लेकिन तुम demo pipeline को कुछ अमान्य parameter इनपुट देकर इसे पहले से ही काम करते हुए देख सकते हो।
3.1.3.1. अपरिचित parameters¶
एक ऐसा parameter पास करने की कोशिश करो जो मौजूद नहीं है:
कंसोल आउटपुट में एक चेतावनी शामिल है:
Pipeline फिर भी चलती है, लेकिन चेतावनी तुम्हें तुरंत सचेत करती है कि --foobar एक मान्यता प्राप्त parameter नहीं है।
यह --outDir जैसी typos को --outdir के बजाय पकड़ता है इससे पहले कि तुम compute समय बर्बाद करो यह सोचते हुए कि आउटपुट गलत जगह क्यों गया।
3.1.3.2. अमान्य parameter values¶
Validation parameter values की भी जांच करता है।
--skip_trim parameter एक boolean flag है, इसलिए एक string value पास करने से pipeline तुरंत fail हो जाती है:
ERROR ~ Validation of pipeline parameters failed!
The following invalid input values have been detected:
* --skip_trim (yes): Value is [string] but should be [boolean]
Pipeline किसी भी process के चलने से पहले रुक जाती है, जिससे तुम एक failed या गलत execution से बच जाते हो।
Boolean parameters को flags के रूप में (--skip_trim) बिना किसी value के पास किया जाना चाहिए, या params फ़ाइल में true/false पर सेट किया जाना चाहिए।
3.1.4. Input validation¶
वही validation logic इनपुट फ़ाइलों की validity जांचने के लिए भी उपयोग किया जा सकता है। उदाहरण के लिए, यदि एक pipeline अपने मुख्य डेटा इनपुट के रूप में एक samplesheet की अपेक्षा करती है (जो कि कई यदि अधिकांश नहीं तो nf-core pipelines का मामला है), तो developer एक input schema (parameters schema से अलग) प्रदान कर सकता है जो बताता है कि इनपुट फ़ाइल कैसे संरचित होनी चाहिए।
फिर, runtime पर, Nextflow जांच कर सकता है कि प्रदान की गई इनपुट फ़ाइल valid है।
हम इसे भाग 5: Input Validation में भी अधिक विस्तार से कवर करते हैं, लेकिन तुम demo pipeline को एक अमान्य input samplesheet देकर इसे पहले से ही काम करते हुए देख सकते हो।
nf-core/demo pipeline sample, fastq_1, और fastq_2 columns के साथ एक CSV फ़ाइल की अपेक्षा करती है।
यह एक schema फ़ाइल (assets/schema_input.json) में परिभाषित है जो अपेक्षित संरचना, column प्रकार और constraints निर्दिष्ट करती है।
assets/schema_input.json
{
"$schema": "https://json-schema.org/draft/2020-12/schema",
"$id": "https://raw.githubusercontent.com/nf-core/demo/master/assets/schema_input.json",
"title": "nf-core/demo pipeline - params.input schema",
"description": "Schema for the file provided with params.input",
"type": "array",
"items": {
"type": "object",
"properties": {
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
"fastq_1": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
},
"fastq_2": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
}
},
"required": ["sample", "fastq_1"]
}
}
Schema निर्दिष्ट करता है कि sample और fastq_1 आवश्यक हैं, जबकि fastq_2 वैकल्पिक है (paired-end और single-end डेटा दोनों का समर्थन करता है)।
File paths को existence और extension pattern के लिए validate किया जाता है।
3.1.4.1. एक अमान्य samplesheet बनाएं¶
एक missing column और एक non-existent file path के साथ एक samplesheet बनाओ:
इस samplesheet में आवश्यक fastq_1 column गायब है और fastq_2 में एक non-existent file path है।
दोनों समस्याएं अगले चरण में validation errors उत्पन्न करेंगी।
3.1.4.2. अमान्य samplesheet के साथ demo pipeline चलाएं¶
malformed_samplesheet.csv को इनपुट के रूप में उपयोग करके demo pipeline चलाओ।
nextflow run nf-core/demo -profile docker,test --outdir demo-results --input malformed_samplesheet.csv
ERROR ~ Validation of pipeline parameters failed!
The following invalid input values have been detected:
* --input (malformed_samplesheet.csv): Validation of file failed:
-> Entry 1: Error for field 'fastq_2' (/not/a/real/file.fastq.gz): the file or directory
'/not/a/real/file.fastq.gz' does not exist (FastQ file for reads 2 cannot contain spaces
and must have extension '.fq.gz' or '.fastq.gz')
-> Entry 1: Missing required field(s): fastq_1
जैसा कि तुम देख सकते हो, pipeline तुरंत fail हो जाती है और सभी validation errors एक साथ रिपोर्ट करती है। nf-schema पहली error पर नहीं रुकता — यह हर समस्या को एकत्र करता है और उन्हें एक साथ सूचीबद्ध करता है, ताकि तुम एक बार में सब कुछ ठीक कर सको बजाय एक-एक करके समस्याओं की खोज करने के।
प्रत्येक error उस exact entry और field की पहचान करती है जिसने समस्या उत्पन्न की, ताकि तुम अपनी samplesheet ठीक कर सको और फिर pipeline को इस विश्वास के साथ फिर से launch कर सको कि यह बाद में किसी बिंदु पर fail नहीं होगी जब Nextflow वास्तव में file path तक पहुँचने जाएगा।
Developers के लिए, यह सब इस पाठ्यक्रम के भाग 5 में अधिक विस्तार से कवर किया गया है।
3.2. Configuration¶
सख्त अर्थ में Configuration नियंत्रित करता है कि pipeline कैसे चलती है: resource allocation, tool-specific arguments, jobs कहाँ execute होते हैं, और कौन सा software packaging system उपयोग करना है।
nf-core pipelines में nextflow.config और conf/ डायरेक्टरी में डिफ़ॉल्ट configuration शामिल है।
कुछ भी override करने से पहले, यह जानना मददगार है कि defaults कहाँ रहते हैं।
तुमने section 2.1 में पहले ही देखा कि pipeline स्रोत कोड $NXF_HOME/assets में रहता है।
उपलब्ध config फ़ाइलें देखने के लिए सूचीबद्ध करो:
सबसे महत्वपूर्ण configuration फ़ाइलें हैं:
conf/base.config: Resource labels (process_low,process_medium,process_high) परिभाषित करता है जो processes को CPUs, memory और time assign करते हैं। जब तुम देखते हो कि एक process अपेक्षा से अधिक resources का उपयोग कर रही है, तो वे defaults यहाँ से आते हैं।conf/modules.config: Per-process tool arguments (ext.args) और output publishing settings (publishDir) सेट करता है। यह फ़ाइल खोलो यह देखने के लिए कि प्रत्येक टूल को डिफ़ॉल्ट रूप से कौन से arguments मिलते हैं।conf/test.config: वह test profile जिसका तुमने section 2.1 में उपयोग किया, जोresourceLimitsके माध्यम से resources को cap करता है और एक test samplesheet सेट करता है।-profile testके साथ activate किया जाता है। पूर्ण आकार के test dataset के साथ चलाने के लिए एकconf/test_full.configभी है, जो benchmarking के लिए उपयोगी है।
केंद्रीय nextflow.config उपरोक्त सभी को load करता है और सब कुछ के लिए उचित डिफ़ॉल्ट मान सेट करता है।
यदि तुम इन फ़ाइलों में निर्दिष्ट किसी भी setting को modify करना चाहते हो, तो इनमें से किसी भी फ़ाइल को सीधे modify मत करो।
इसके बजाय, अपनी खुद की config फ़ाइल बनाओ और इसे -c के साथ पास करो।
तुम्हारे द्वारा निर्दिष्ट values उन अन्य फ़ाइलों में सेट किए गए डिफ़ॉल्ट values को override करेंगी।
आइए इसे व्यवहार में करने के लिए कुछ अभ्यास करें।
3.2.1. एक process के लिए resource allocation बदलें¶
Demo pipeline base.config में परिभाषित labels का उपयोग करके resources assign करती है।
उदाहरण के लिए, FASTQC process_medium label का उपयोग करता है, जो 6 CPUs और 36 GB memory allocate करता है।
Test profile resourceLimits के माध्यम से resources को cap करता है, लेकिन तुम specific processes के लिए resources को भी override कर सकते हो।
custom.config नामक एक फ़ाइल बनाओ:
अपने custom config के साथ pipeline चलाओ:
कमांड आउटपुट
-c flag तुम्हारे config को pipeline के built-in configuration के ऊपर जोड़ता है।
3.2.2. ext.args के साथ tool argument values सेट करें¶
कई command-line tools में ऐसे arguments होते हैं जो आवश्यक नहीं होते और इसलिए pipeline parameters के रूप में सेट नहीं किए जाते जब तक कि वे बहुत सामान्य रूप से उपयोग न किए जाएं।
उन tool arguments के लिए, nf-core modules एक Nextflow convention का उपयोग करते हैं जिसे ext.args कहा जाता है जो एक configuration फ़ाइल के माध्यम से underlying tool को arguments पास करता है।
उदाहरण के लिए, आइए ext.args का उपयोग करके SEQTK_TRIM module में एक trimming argument जोड़ें।
3.2.2.1. Custom configuration को update करें¶
अपना custom.config update करो:
| custom.config | |
|---|---|
यह seqtk trimfq को quality trimming के अलावा प्रत्येक read की शुरुआत से 5 bases trim करने के लिए कहता है।
3.2.2.2. Pipeline चलाएं¶
इस config के साथ pipeline को फिर से चलाओ प्रभाव देखने के लिए:
कमांड आउटपुट
यह verify करने के लिए कि argument apply किया गया था, run output से SEQTK_TRIM work directory hash खोजो (जैसे work/ab/cd1234...) और उसके अंदर .command.sh फ़ाइल जांचो:
कमांड आउटपुट
तुम्हें seqtk trimfq कमांड में -b 5 दिखना चाहिए, जो पुष्टि करता है कि तुम्हारा ext.args override प्रभावी हुआ।
3.2.2.3. डिफ़ॉल्ट values को override करना¶
कुछ modules में ext.args पहले से डिफ़ॉल्ट रूप से सेट होता है।
उदाहरण के लिए, FASTQC module डिफ़ॉल्ट रूप से ext.args = '--quiet' के साथ configure किया गया है (conf/modules.config में परिभाषित)।
| conf/modules.config | |
|---|---|
यदि तुम एक custom configuration फ़ाइल के माध्यम से ext.args के लिए एक value प्रदान करते हो, तो वह value उस process के लिए सेट किए गए डिफ़ॉल्ट को पूरी तरह से replace कर देगी।
इसलिए उदाहरण के लिए, यदि डिफ़ॉल्ट '--quiet' था और तुम ext.args = '--kmers 8' सेट करते हो, तो --quiet flag अब apply नहीं होगा।
दोनों को रखने के लिए, ext.args = '--quiet --kmers 8' सेट करो।
इसका मतलब है कि तुम उन tools के डिफ़ॉल्ट configuration की जांच करने के लिए जिम्मेदार हो जिन्हें तुम ext.args के साथ argument values प्रदान करना चाहते हो।
सारांश¶
तुम जानते हो कि एक nf-core pipeline से help कैसे प्राप्त करें, parameters कैसे सेट करें और यह समझें कि उन्हें कैसे validate किया जाता है, और config फ़ाइलों के माध्यम से configuration को कैसे customize करें।
आगे क्या है?¶
एक ब्रेक लो! जब तुम तैयार हो, तो भाग 2 पर जाओ, जहाँ तुम scratch से अपनी खुद की nf-core compatible pipeline बनाओगे।