Parte 1: Eseguire una pipeline demo¶
Traduzione assistita da IA - scopri di più e suggerisci miglioramenti
In questa prima parte del corso di formazione Hello nf-core, mostreremo come trovare e provare una pipeline nf-core, configurare e personalizzare la sua esecuzione in base alle proprie esigenze, e capire come la validazione dell'input protegge dagli errori più comuni.
Utilizzeremo una pipeline chiamata nf-core/demo che è mantenuta dal progetto nf-core come parte del suo inventario di pipeline per scopi dimostrativi e di formazione.
Assicuratevi che la vostra directory di lavoro sia impostata su hello-nf-core/ come indicato nella pagina Iniziare.
1. Trovare e recuperare la pipeline nf-core/demo¶
Iniziamo localizzando la pipeline nf-core/demo sul sito web del progetto nf-co.re, che centralizza tutte le informazioni come: documentazione generale e articoli di aiuto, documentazione per ciascuna delle pipeline, post di blog, annunci di eventi e così via.
1.1. Trovare la pipeline sul sito web¶
Nel vostro browser web, andate su https://nf-co.re/pipelines/ e digitate demo nella barra di ricerca.
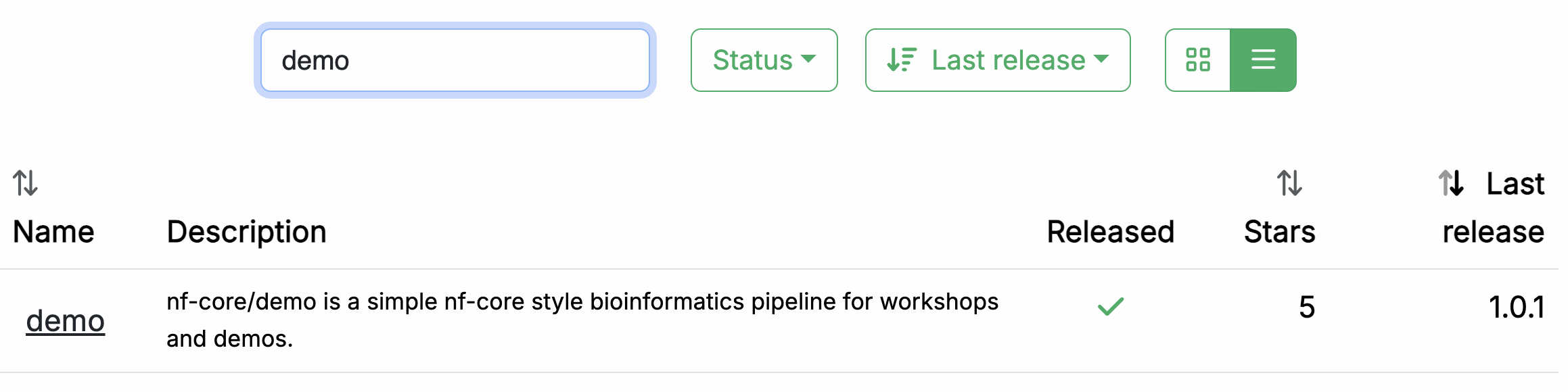
Cliccate sul nome della pipeline, demo, per accedere alla pagina di documentazione della pipeline.
Ogni pipeline rilasciata ha una pagina dedicata che include le seguenti sezioni di documentazione:
- Introduction: Un'introduzione e panoramica della pipeline
- Usage: Descrizioni di come eseguire la pipeline
- Parameters: Parametri della pipeline raggruppati con descrizioni
- Output: Descrizioni ed esempi dei file di output previsti
- Results: File di output di esempio generati dal dataset di test completo
- Releases & Statistics: Cronologia delle versioni della pipeline e statistiche
Quando state considerando di adottare una nuova pipeline, dovreste leggere attentamente la documentazione della pipeline prima per comprendere cosa fa e come dovrebbe essere configurata prima di tentare di eseguirla.
Date un'occhiata ora e vedete se riuscite a scoprire:
- Quali strumenti la pipeline eseguirà (Controllate la scheda:
Introduction) - Quali input e parametri la pipeline accetta o richiede (Controllate la scheda:
Parameters) - Quali sono gli output prodotti dalla pipeline (Controllate la scheda:
Output)
1.1.1. Panoramica della pipeline¶
La scheda Introduction fornisce una panoramica della pipeline, inclusa una rappresentazione visiva (chiamata mappa della metropolitana) e un elenco di strumenti che vengono eseguiti come parte della pipeline.

- Read QC (FASTQC)
- Adapter and quality trimming (SEQTK_TRIM)
- Present QC for raw reads (MULTIQC)
1.1.2. Esempio di riga di comando¶
La documentazione fornisce anche un file di input di esempio (discusso ulteriormente più avanti) e un esempio di riga di comando.
nextflow run nf-core/demo \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>
Noterete che il comando di esempio NON specifica un file workflow, solo il riferimento al repository della pipeline, nf-core/demo.
Quando invocato in questo modo, Nextflow assumerà che il codice sia organizzato in un certo modo. Recuperiamo il codice così possiamo esaminare questa struttura.
1.2. Recuperare il codice della pipeline¶
Una volta determinato che la pipeline sembra essere adatta ai nostri scopi, proviamola. Fortunatamente Nextflow rende facile recuperare pipeline da repository formattati correttamente senza dover scaricare nulla manualmente.
1.2.1. Usare nextflow pull¶
Torniamo al terminale ed eseguiamo quanto segue:
Output del comando
Nextflow esegue un pull del codice della pipeline, cioè scarica il repository completo sul vostro disco locale.
Per essere chiari, potete farlo con qualsiasi pipeline Nextflow che sia configurata appropriatamente in GitHub, non solo le pipeline nf-core. Tuttavia nf-core è la più grande collezione open-source di pipeline Nextflow.
1.2.2. Usare nextflow list¶
Potete ottenere da Nextflow un elenco di quali pipeline avete recuperato in questo modo:
Potete provare a fare il pull di alcune altre pipeline per vedere come vengono elencate quando ne avete più di una.
1.2.3. Trovare le pipeline in $NXF_HOME/assets/¶
Noterete che i file non sono nella vostra directory di lavoro corrente.
Per impostazione predefinita, Nextflow li salva in $NXF_HOME/assets.
Nota
Il percorso completo potrebbe differire sul vostro sistema se non state utilizzando il nostro ambiente di formazione.
Nextflow mantiene intenzionalmente il codice sorgente scaricato 'fuori mano' sul principio che queste pipeline dovrebbero essere utilizzate più come librerie che come codice con cui interagire direttamente.
1.2.4. Creare un symlink per accedere facilmente al codice sorgente¶
Non esamineremo il codice in dettaglio, ma diamo una rapida occhiata per farci un'idea di come appare l'organizzazione generale.
Per rendere più facile sfogliare il codice sorgente della pipeline, create un collegamento simbolico alla directory degli asset:
Questo crea una scorciatoia che vi permette di esplorare il codice con tree -L 2 pipelines o di aprire i file direttamente.
1.2.5. Panoramica dell'organizzazione del codice¶
Potete usare tree o utilizzare l'esploratore di file per trovare e aprire la directory nf-core/demo.
Contenuto della directory
pipelines/nf-core/demo
├── assets
├── CHANGELOG.md
├── CITATIONS.md
├── CODE_OF_CONDUCT.md
├── conf
├── docs
├── LICENSE
├── main.nf
├── modules
├── modules.json
├── nextflow.config
├── nextflow_schema.json
├── nf-test.config
├── README.md
├── ro-crate-metadata.json
├── subworkflows
├── tests
├── tower.yml
└── workflows
Come potete vedere, c'è molto in corso là dentro, ma la maggior parte non richiede la vostra attenzione.
In breve, notiamo che al livello superiore potete trovare un file README con informazioni di riepilogo, così come file accessori che riassumono informazioni sul progetto come licenza, linee guida per i contributi, citazioni e codice di condotta.
La documentazione dettagliata della pipeline si trova nella directory docs.
Tutto questo contenuto viene utilizzato per generare le pagine web sul sito web nf-core in modo programmatico, quindi sono sempre aggiornate con il codice.
Per il resto, possiamo distinguere tre gruppi funzionali di file di codice:
- Componenti del codice della pipeline (
main.nf,workflows,subworkflows,modules) - Configurazione della pipeline
- Parametri della pipeline / input e validazione
Non esamineremo i componenti del codice della pipeline in questa parte del corso, ma toccheremo gli elementi di configurazione e validazione che potrebbero essere rilevanti per voi come utenti finali delle pipeline nf-core.
Suggerimento
Potete anche sfogliare il codice sorgente di qualsiasi pipeline nf-core su GitHub, ad esempio github.com/nf-core/demo. Ogni pipeline nf-core segue la stessa struttura di directory, quindi una volta che conoscete la struttura, potete trovare file di configurazione, moduli e workflow per qualsiasi pipeline nello stesso modo.
Ma per ora, passiamo all'esecuzione della pipeline!
Takeaway¶
Ora sapete come trovare una pipeline tramite il sito web nf-core e recuperare una copia locale del codice sorgente.
Cosa c'è dopo?¶
Imparate come provare una pipeline nf-core con il minimo sforzo.
2. Provare la pipeline con il suo profilo di test¶
Convenientemente, ogni pipeline nf-core viene fornita con un profilo di test. Questo è un set minimo di impostazioni di configurazione per l'esecuzione della pipeline utilizzando un piccolo dataset di test ospitato nel repository nf-core/test-datasets. È un ottimo modo per provare rapidamente una pipeline su piccola scala.
Nota
Il sistema di profili di configurazione di Nextflow vi permette di passare facilmente tra diversi motori di container o ambienti di esecuzione. Per maggiori dettagli, vedete Hello Nextflow Parte 6: Configuration.
2.1. Esaminare il profilo di test¶
È buona pratica verificare cosa specifica il profilo di test di una pipeline prima di eseguirla.
Il profilo test per nf-core/demo risiede nel file di configurazione conf/test.config.
Potete trovarlo localmente all'interno del codice sorgente della pipeline scaricato da nextflow pull:
Ecco il contenuto di quel file:
Noterete subito che il blocco di commenti in alto include un esempio di utilizzo che mostra come eseguire la pipeline con questo profilo di test.
| conf/test.config | |
|---|---|
Le uniche cose che dobbiamo fornire sono quelle mostrate tra parentesi angolari nell'esempio di comando: <docker/singularity> e <OUTDIR>.
Come promemoria, <docker/singularity> si riferisce alla scelta del sistema di container. Tutte le pipeline nf-core sono progettate per essere utilizzabili con container (Docker, Singularity, ecc.) per garantire la riproducibilità ed eliminare problemi di installazione del software.
Quindi dovremo specificare se vogliamo usare Docker o Singularity per testare la pipeline.
La parte --outdir <OUTDIR> si riferisce alla directory in cui Nextflow scriverà gli output della pipeline.
Dobbiamo fornire un nome per essa, che possiamo semplicemente inventare.
Se non esiste già, Nextflow la creerà per noi durante l'esecuzione.
Passando alla sezione dopo il blocco di commenti, il profilo di test ci mostra cosa è stato preconfigurato per il test: in particolare, il parametro input è già impostato per puntare a un dataset di test, quindi non dobbiamo fornire i nostri dati.
Se seguite il link all'input preconfigurato, vedrete che è un file CSV contenente identificatori di campioni e percorsi di file per diversi campioni sperimentali.
sample,fastq_1,fastq_2
SAMPLE1_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R2.fastq.gz
SAMPLE2_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R2.fastq.gz
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,
Questo è chiamato samplesheet, ed è la forma più comune di input per le pipeline nf-core.
Nota
Non preoccupatevi se non avete familiarità con i formati e i tipi di dati, non è importante per quello che segue.
Quindi questo conferma che abbiamo tutto ciò di cui abbiamo bisogno per provare la pipeline.
2.2. Eseguire la pipeline¶
Decidiamo di usare Docker per il sistema di container e demo-results come directory di output, e siamo pronti per eseguire il comando di test:
Output del comando
N E X T F L O W ~ version 25.10.4
Launching `https://github.com/nf-core/demo` [magical_pauling] DSL2 - revision: 45904cb9d1 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.1.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2025-11-21_04-57-41
Core Nextflow options
revision : master
runName : magical_pauling
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/nf-core/demo
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/nf-core/demo/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (7)
[ff/a6976b] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) | 3 of 3 ✔
[39/731ab7] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) | 3 of 3 ✔
[7c/78d96e] NFCORE_DEMO:DEMO:MULTIQC | 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Se il vostro output corrisponde a quello, congratulazioni! Avete appena eseguito la vostra prima pipeline nf-core.
Noterete che c'è molto più output sulla console rispetto a quando eseguite una pipeline Nextflow di base. C'è un'intestazione che include un riepilogo della versione della pipeline, input e output, e alcuni elementi di configurazione.
Nota
Il vostro output mostrerà timestamp, nomi di esecuzione e percorsi di file diversi, ma la struttura complessiva e l'esecuzione dei processi dovrebbero essere simili.
Notate la riga vicino alla cima dell'output:
Questo vi dice quale revisione della pipeline è stata utilizzata.
Poiché non abbiamo specificato una versione, Nextflow ha usato l'ultimo commit su master.
Per esecuzioni riproducibili, dovreste fissare una release specifica usando il flag -r:
Questo garantisce che lo stesso codice della pipeline venga utilizzato ogni volta, indipendentemente da nuovi commit o release.
Per questa formazione omettiamo -r per semplicità, ma in produzione dovreste sempre specificarlo.
Passando all'output di esecuzione, diamo un'occhiata alle righe che ci dicono quali processi sono stati eseguiti:
executor > local (7)
[ff/a6976b] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) | 3 of 3 ✔
[39/731ab7] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) | 3 of 3 ✔
[7c/78d96e] NFCORE_DEMO:DEMO:MULTIQC | 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Questo ci dice che sono stati eseguiti tre processi, corrispondenti ai tre strumenti mostrati nella pagina di documentazione della pipeline sul sito web nf-core: FASTQC, SEQTK_TRIM e MULTIQC.
I nomi completi dei processi come mostrati qui, come NFCORE_DEMO:DEMO:MULTIQC, sono più lunghi di quelli che potreste aver visto nel materiale introduttivo Hello Nextflow.
Questi includono i nomi dei loro workflow padre e riflettono la modularità del codice della pipeline.
Entreremo più nel dettaglio nella Parte 2 di questo corso.
2.3. Esaminare gli output della pipeline¶
Infine, diamo un'occhiata alla directory demo-results prodotta dalla pipeline.
Contenuto della directory
demo-results
├── fastqc
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── fq
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── multiqc
│ ├── multiqc_data
│ ├── multiqc_plots
│ └── multiqc_report.html
└── pipeline_info
├── execution_report_2025-11-21_04-57-41.html
├── execution_timeline_2025-11-21_04-57-41.html
├── execution_trace_2025-11-21_04-57-41.txt
├── nf_core_demo_software_mqc_versions.yml
├── params_2025-11-21_04-57-46.json
└── pipeline_dag_2025-11-21_04-57-41.html
Potrebbe sembrare molto.
Per saperne di più sugli output della pipeline nf-core/demo, consultate la sua pagina di documentazione.
In questa fase, ciò che è importante osservare è che i risultati sono organizzati per modulo, e c'è inoltre una directory chiamata pipeline_info contenente vari report con timestamp sull'esecuzione della pipeline.
Per esempio, il file execution_timeline_* vi mostra quali processi sono stati eseguiti, in quale ordine e quanto tempo hanno impiegato per essere eseguiti:
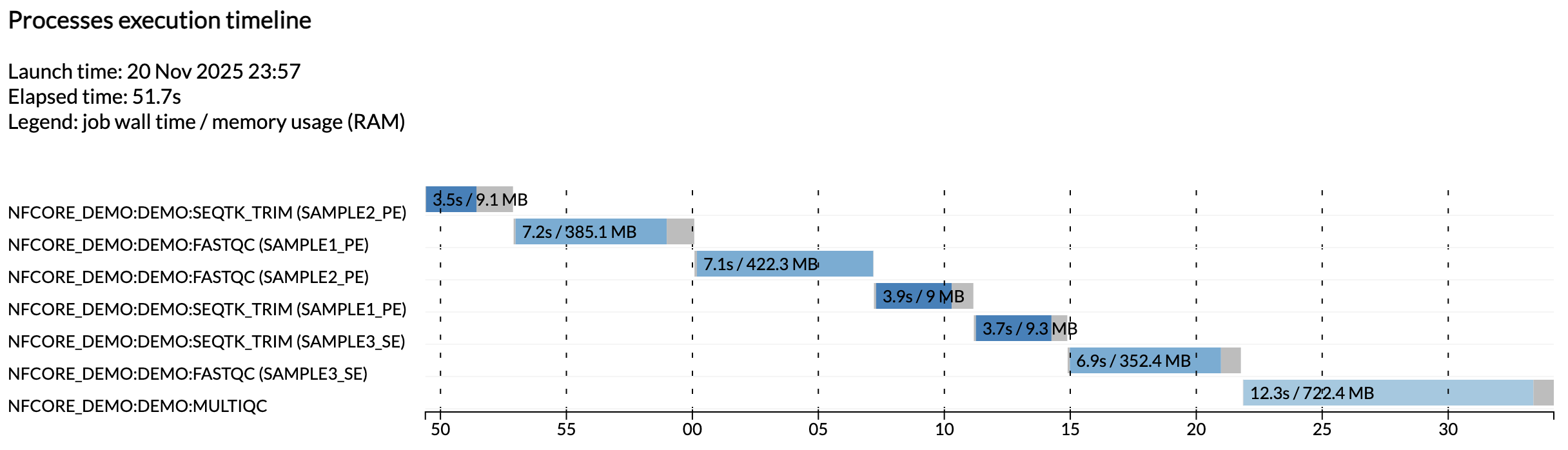
Nota
Qui le attività non sono state eseguite in parallelo perché stiamo eseguendo su una macchina minimalista in Github Codespaces. Per vedere queste eseguite in parallelo, provate ad aumentare l'allocazione CPU del vostro codespace e i limiti di risorse nella configurazione di test.
Questi report sono generati automaticamente per tutte le pipeline nf-core.
Takeaway¶
Sapete come eseguire una pipeline nf-core utilizzando il suo profilo di test integrato e dove trovare i suoi output.
Cosa c'è dopo?¶
Imparate come configurare la pipeline per personalizzare la sua esecuzione.
3. Configurare l'esecuzione della pipeline¶
Come spiegato in Hello Config, vogliamo poter cambiare su quali dati la nostra pipeline verrà eseguita e come verrà eseguita senza modificare il codice della pipeline stesso. A tal fine, Nextflow supporta diversi modi per controllare la configurazione della pipeline, il che può risultare un po' opprimente.
Il progetto nf-core specifica convenzioni per organizzare gli elementi di configurazione, distinguendo due tipi di configurazione al livello superiore: parametri della pipeline e configurazione in senso stretto.
- Parametri della pipeline (impostati tramite il sistema
params) includono tipicamente cose come file di input, flag di comportamento degli strumenti e parametri di analisi. - Configurazione in senso stretto si riferisce alla logistica di come la pipeline viene eseguita, cioè l'executor, le allocazioni di risorse di calcolo e così via.
Iniziamo affrontando i parametri della pipeline, poi esamineremo la configurazione in senso stretto.
3.1. Parametri della pipeline¶
Per tutte le pipeline nf-core, potete ottenere un elenco completo dei parametri della pipeline direttamente dalla riga di comando usando il flag --help, che è esso stesso un parametro della pipeline.
3.1.1. Ottenere l'elenco dei parametri con --help¶
Eseguite il comando di aiuto per la pipeline demo:
Output del comando
N E X T F L O W ~ version 25.10.4
Launching `https://github.com/nf-core/demo` [run_name] DSL2 - revision: 45904cb9d1 [master]
----------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.1.0
----------------------------------------------------
Typical pipeline command:
nextflow run nf-core/demo -profile <docker/singularity/.../institute> --input samplesheet.csv --outdir <OUTDIR>
Input/output options
--input [string] Path to a metadata file containing information about the samples in the experiment.
--outdir [string] The output directory where the results will be saved. You have to use absolute paths to storage on Cloud infrastructure.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header, used for filename if not otherwise specified.
Reference genome options
--genome [string] Name of iGenomes reference.
--fasta [string] Path to FASTA genome file.
Process skipping options
--skip_trim [boolean] Skip trimming fastq files with seqtk
Generic options
--multiqc_methods_description [string] Custom MultiQC yaml file containing HTML including a methods description.
--help [boolean, string] Display the help message.
--help_full [boolean] Display the full detailed help message.
--show_hidden [boolean] Display hidden parameters in the help message (only works when --help or --help_full are provided).
!! Hiding 20 param(s), use the `--show_hidden` parameter to show them !!
----------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
Come potete vedere, l'output raggruppa i parametri in categorie (opzioni di input/output, opzioni del genoma di riferimento, ecc.) con tipi e descrizioni per ciascuno.
Questa categorizzazione è determinata da un file di schema, di cui parleremo più avanti.
Nelle pipeline Nextflow semplici, --help funziona solo se lo sviluppatore lo ha implementato manualmente.
Suggerimento
Usate --help --show_hidden per vedere i parametri aggiuntivi che sono nascosti per impostazione predefinita, come --publish_dir_mode o --monochrome_logs.
3.1.2. Impostare i valori dei parametri¶
Come trattato in Hello Config, potete impostare i valori dei parametri dalla riga di comando con --nome_parametro o raccogliere un insieme di parametri in un file YAML e passarlo con -params-file.
Entrambi gli approcci funzionano allo stesso modo con le pipeline nf-core.
Per esempio, per saltare la fase di trimming:
Output del comando
Il processo SEQTK_TRIM non appare più nell'output.
Info
Sebbene sia tecnicamente possibile impostare i parametri della pipeline in un file di configurazione personalizzato passato con -c, questo potrebbe non sovrascrivere i valori predefiniti già impostati nel nextflow.config della pipeline, a seconda delle regole di precedenza della configurazione di Nextflow.
Usare --nome_parametro dalla riga di comando o -params-file è più affidabile, poiché questi hanno sempre la precedenza.
Come regola generale: se appare nell'output di --help, impostatelo tramite la riga di comando o un file di parametri piuttosto che un file di configurazione.
3.1.3. Validazione dei parametri¶
Curiosità: il comando --help funziona per tutte le pipeline nf-core perché il progetto nf-core richiede agli sviluppatori di definire formalmente tutti i parametri della pipeline in un file di schema JSON (nextflow_schema.json).
Questo schema registra il tipo, la descrizione, il valore predefinito e il raggruppamento di ciascun parametro.
Oltre a supportare l'output di --help, il file di schema abilita anche la validazione automatizzata al momento del lancio.
Ciò significa che Nextflow può verificare che ogni parametro passato esista e abbia ricevuto un valore appropriato (del tipo appropriato, nell'intervallo di valori consentito, ecc.).
Tratteremo questo in maggior dettaglio nella Parte 5: Input Validation, ma potete già vederlo in azione fornendo alla pipeline demo un input di parametri non valido.
3.1.3.1. Parametri non riconosciuti¶
Provate a passare un parametro che non esiste:
L'output della console include un avviso:
La pipeline continua a essere eseguita, ma l'avviso vi avvisa immediatamente che --foobar non è un parametro riconosciuto.
Questo intercetta errori di battitura come --outDir invece di --outdir prima che sprechiate tempo di calcolo chiedendovi perché l'output è finito nel posto sbagliato.
3.1.3.2. Valori di parametri non validi¶
La validazione controlla anche i valori dei parametri.
Il parametro --skip_trim è un flag boolean, quindi passare un valore stringa causa il fallimento immediato della pipeline:
ERROR ~ Validation of pipeline parameters failed!
The following invalid input values have been detected:
* --skip_trim (yes): Value is [string] but should be [boolean]
La pipeline si ferma prima che vengano eseguiti i processi, risparmiandovi un'esecuzione fallita o errata.
I parametri boolean dovrebbero essere passati come flag (--skip_trim) senza un valore, oppure impostati a true/false in un file di parametri.
3.1.4. Validazione dell'input¶
La stessa logica di validazione può essere utilizzata anche per verificare la validità dei file di input. Per esempio, se una pipeline si aspetta un samplesheet come input principale dei dati (il che è il caso di molte se non della maggior parte delle pipeline nf-core), lo sviluppatore può fornire uno schema di input (distinto dallo schema dei parametri) che descrive come il file di input dovrebbe essere strutturato.
Poi, durante l'esecuzione, Nextflow può verificare che il file di input fornito sia valido.
Tratteremo anche questo in maggior dettaglio nella Parte 5: Input Validation, ma potete già vederlo in azione fornendo alla pipeline demo un samplesheet di input non valido.
La pipeline nf-core/demo si aspetta un file CSV con le colonne sample, fastq_1 e fastq_2.
Questo è definito in un file di schema (assets/schema_input.json) che specifica la struttura attesa, i tipi di colonna e i vincoli.
assets/schema_input.json
{
"$schema": "https://json-schema.org/draft/2020-12/schema",
"$id": "https://raw.githubusercontent.com/nf-core/demo/master/assets/schema_input.json",
"title": "nf-core/demo pipeline - params.input schema",
"description": "Schema for the file provided with params.input",
"type": "array",
"items": {
"type": "object",
"properties": {
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
"fastq_1": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
},
"fastq_2": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
}
},
"required": ["sample", "fastq_1"]
}
}
Lo schema specifica che sample e fastq_1 sono obbligatori, mentre fastq_2 è opzionale (supportando sia dati paired-end che single-end).
I percorsi dei file vengono validati per esistenza e pattern dell'estensione.
3.1.4.1. Creare un samplesheet non valido¶
Create un samplesheet con una colonna mancante e un percorso di file inesistente:
Questo samplesheet manca della colonna obbligatoria fastq_1 e ha un percorso di file inesistente in fastq_2.
Entrambi i problemi produrranno errori di validazione nel passo successivo.
3.1.4.2. Eseguire la pipeline demo con il samplesheet non valido¶
Eseguite la pipeline demo usando malformed_samplesheet.csv come input.
nextflow run nf-core/demo -profile docker,test --outdir demo-results --input malformed_samplesheet.csv
ERROR ~ Validation of pipeline parameters failed!
The following invalid input values have been detected:
* --input (malformed_samplesheet.csv): Validation of file failed:
-> Entry 1: Error for field 'fastq_2' (/not/a/real/file.fastq.gz): the file or directory
'/not/a/real/file.fastq.gz' does not exist (FastQ file for reads 2 cannot contain spaces
and must have extension '.fq.gz' or '.fastq.gz')
-> Entry 1: Missing required field(s): fastq_1
Come potete vedere, la pipeline fallisce immediatamente e riporta tutti gli errori di validazione in una volta sola. nf-schema non si ferma al primo errore — raccoglie ogni problema e li elenca insieme, così potete correggere tutto in una volta sola invece di scoprire i problemi uno alla volta.
Ogni errore identifica la voce e il campo esatti che hanno causato il problema, così potete correggere il vostro samplesheet e poi rilanciare la pipeline con la certezza che non fallirà in un momento successivo quando Nextflow andrà effettivamente ad accedere al percorso del file.
Per gli sviluppatori, tutto questo è trattato in maggior dettaglio nella Parte 5 di questo corso.
3.2. Configurazione¶
La configurazione in senso stretto controlla come viene eseguita la pipeline: allocazione delle risorse, argomenti specifici degli strumenti, dove vengono eseguiti i job e quale sistema di packaging del software utilizzare.
Le pipeline nf-core includono la configurazione predefinita in nextflow.config e nella directory conf/.
Prima di sovrascrivere qualsiasi cosa, è utile sapere dove si trovano i valori predefiniti.
Avete già visto nella sezione 2.1 che il codice sorgente della pipeline si trova in $NXF_HOME/assets.
Elencate i file di configurazione per vedere cosa è disponibile:
I file di configurazione più importanti sono:
conf/base.config: Definisce le etichette delle risorse (process_low,process_medium,process_high) che assegnano CPU, memoria e tempo ai processi. Quando vedete un processo che utilizza più risorse del previsto, è qui che si trovano quei valori predefiniti.conf/modules.config: Imposta gli argomenti degli strumenti per processo (ext.args) e le impostazioni di pubblicazione dell'output (publishDir). Aprite questo file per vedere quali argomenti riceve ogni strumento per impostazione predefinita.conf/test.config: Il profilo di test che avete usato nella sezione 2.1, che limita le risorse tramiteresourceLimitse imposta un samplesheet di test. Attivato con-profile test. Esiste anche unconf/test_full.configper l'esecuzione con un dataset di test di dimensioni complete, utile per il benchmarking.
Il nextflow.config centrale carica tutti i file sopra indicati e imposta i valori predefiniti appropriati per tutto.
Se desiderate modificare qualsiasi impostazione specificata in questi file, non modificate nessuno di essi direttamente.
Create invece il vostro file di configurazione e passatelo con -c.
I valori che specificate sovrascriveranno i valori predefiniti impostati in quegli altri file.
Eseguiamo alcuni esercizi per farlo in pratica.
3.2.1. Modificare l'allocazione delle risorse per un processo¶
La pipeline demo assegna le risorse usando etichette definite in base.config.
Per esempio, FASTQC usa l'etichetta process_medium, che alloca 6 CPU e 36 GB di memoria.
Il profilo di test limita le risorse tramite resourceLimits, ma potete anche sovrascrivere le risorse per processi specifici.
Create un file chiamato custom.config:
Eseguite la pipeline con la vostra configurazione personalizzata:
Output del comando
Il flag -c aggiunge la vostra configurazione sopra la configurazione integrata della pipeline.
3.2.2. Impostare i valori degli argomenti degli strumenti con ext.args¶
Molti strumenti da riga di comando hanno argomenti che non sono obbligatori e quindi non vengono impostati come parametri della pipeline a meno che non siano molto comunemente usati.
Per quegli argomenti degli strumenti, i moduli nf-core usano una convenzione Nextflow chiamata ext.args per passare argomenti allo strumento sottostante tramite un file di configurazione.
Per esempio, aggiungiamo un argomento di trimming al modulo SEQTK_TRIM usando ext.args.
3.2.2.1. Aggiornare la configurazione personalizzata¶
Aggiornate il vostro custom.config:
| custom.config | |
|---|---|
Questo dice a seqtk trimfq di tagliare 5 basi dall'inizio di ogni read in aggiunta al trimming di qualità.
3.2.2.2. Eseguire la pipeline¶
Eseguite di nuovo la pipeline con questa configurazione per vedere l'effetto:
Output del comando
Per verificare che l'argomento sia stato applicato, trovate l'hash della directory di lavoro di SEQTK_TRIM dall'output dell'esecuzione (ad es. work/ab/cd1234...) e controllate il file .command.sh al suo interno:
Output del comando
Dovreste vedere -b 5 nel comando seqtk trimfq, confermando che la vostra sovrascrittura di ext.args ha avuto effetto.
3.2.2.3. Sovrascrivere i valori predefiniti¶
Alcuni moduli hanno ext.args già impostato per impostazione predefinita.
Per esempio, il modulo FASTQC è configurato con ext.args = '--quiet' per impostazione predefinita (definito in conf/modules.config).
| conf/modules.config | |
|---|---|
Se fornite un valore per ext.args tramite un file di configurazione personalizzato, quel valore sostituirà completamente il valore predefinito impostato per quel processo.
Quindi per esempio, se il valore predefinito era '--quiet' e impostate ext.args = '--kmers 8', il flag --quiet non verrà più applicato.
Per mantenere entrambi, impostate ext.args = '--quiet --kmers 8'.
Questo significa che siete responsabili di verificare qual è la configurazione predefinita degli strumenti a cui volete fornire valori di argomenti con ext.args.
Takeaway¶
Sapete come ottenere aiuto da una pipeline nf-core, impostare i parametri e capire come vengono validati, e personalizzare la configurazione tramite file di configurazione.
Cosa c'è dopo?¶
Prendetevi una pausa! Quando siete pronti, passate alla Parte 2, dove creerete la vostra pipeline compatibile con nf-core da zero.