Część 1: Uruchomienie demonstracyjnego pipeline'a¶
Tłumaczenie wspomagane przez AI - dowiedz się więcej i zasugeruj ulepszenia
W tej pierwszej części szkolenia Hello nf-core pokażemy Ci, jak znaleźć i wypróbować pipeline nf-core, skonfigurować i dostosować jego wykonanie do swoich potrzeb oraz zrozumieć, jak walidacja wejścia chroni przed typowymi błędami.
Będziemy używać pipeline'a o nazwie nf-core/demo, który jest utrzymywany przez projekt nf-core jako część inwentarza pipeline'ów służących do demonstracji i celów szkoleniowych.
Upewnij się, że Twój katalog roboczy jest ustawiony na hello-nf-core/, jak wskazano na stronie Pierwsze kroki.
1. Znajdź i pobierz pipeline nf-core/demo¶
Zacznijmy od zlokalizowania pipeline'a nf-core/demo na stronie projektu pod adresem nf-co.re, która centralizuje wszystkie informacje, takie jak: ogólna dokumentacja i artykuły pomocnicze, dokumentacja dla każdego z pipeline'ów, wpisy na blogu, ogłoszenia wydarzeń i tak dalej.
1.1. Znajdź pipeline na stronie internetowej¶
W przeglądarce internetowej przejdź do https://nf-co.re/pipelines/ i wpisz demo w pasku wyszukiwania.
Kliknij nazwę pipeline'a, demo, aby uzyskać dostęp do strony dokumentacji pipeline'a.
Każdy wydany pipeline ma dedykowaną stronę zawierającą następujące sekcje dokumentacji:
- Introduction: Wprowadzenie i przegląd pipeline'a
- Usage: Opisy sposobu wykonywania pipeline'a
- Parameters: Zgrupowane parametry pipeline'a z opisami
- Output: Opisy i przykłady oczekiwanych plików wyjściowych
- Results: Przykładowe pliki wyjściowe wygenerowane z pełnego zestawu danych testowych
- Releases & Statistics: Historia wersji pipeline'a i statystyki
Gdy rozważasz przyjęcie nowego pipeline'a, powinieneś najpierw uważnie przeczytać jego dokumentację, aby zrozumieć, co robi i jak powinien być skonfigurowany, zanim spróbujesz go uruchomić.
Spójrz teraz i zobacz, czy możesz dowiedzieć się:
- Które narzędzia uruchomi pipeline (Sprawdź zakładkę:
Introduction) - Jakie wejścia i parametry pipeline akceptuje lub wymaga (Sprawdź zakładkę:
Parameters) - Jakie są wyjścia produkowane przez pipeline (Sprawdź zakładkę:
Output)
1.1.1. Przegląd pipeline'a¶
Zakładka Introduction dostarcza przegląd pipeline'a, w tym wizualną reprezentację (zwaną mapą metra) i listę narzędzi uruchamianych jako część pipeline'a.

- Read QC (FASTQC)
- Adapter and quality trimming (SEQTK_TRIM)
- Present QC for raw reads (MULTIQC)
1.1.2. Przykładowa linia poleceń¶
Dokumentacja dostarcza również przykładowy plik wejściowy (omówiony dokładniej poniżej) i przykładową linię poleceń.
nextflow run nf-core/demo \
-profile <docker/singularity/.../institute> \
--input samplesheet.csv \
--outdir <OUTDIR>
Zauważysz, że przykładowe polecenie NIE określa pliku workflow'u, tylko referencję do repozytorium pipeline'a, nf-core/demo.
Gdy jest wywoływany w ten sposób, Nextflow zakłada, że kod jest zorganizowany w określony sposób. Pobierzmy kod, abyśmy mogli zbadać tę strukturę.
1.2. Pobierz kod pipeline'a¶
Gdy ustaliliśmy już, że pipeline wydaje się być odpowiedni dla naszych celów, wypróbujmy go. Na szczęście Nextflow ułatwia pobieranie pipeline'ów z poprawnie sformatowanych repozytoriów bez konieczności ręcznego pobierania czegokolwiek.
1.2.1. Użyj nextflow pull¶
Wróćmy do terminala i uruchommy następujące polecenie:
Wyjście polecenia
Nextflow wykonuje pull kodu pipeline'a, co oznacza, że pobiera całe repozytorium na Twój dysk lokalny.
Wyjaśnijmy, że możesz to zrobić z każdym workflow'em Nextflow, który jest odpowiednio skonfigurowany w GitHub, nie tylko z pipeline'ami nf-core. Jednak nf-core to największa otwarta kolekcja workflow'ów Nextflow.
1.2.2. Użyj nextflow list¶
Możesz uzyskać od Nextflow listę pipeline'ów, które pobrałeś w ten sposób:
Możesz spróbować pobrać kilka innych pipeline'ów, aby zobaczyć, jak są wyświetlane, gdy masz ich więcej niż jeden.
1.2.3. Znajdź swoje pipeline'y w $NXF_HOME/assets/¶
Zauważysz, że pliki nie znajdują się w Twoim bieżącym katalogu roboczym.
Domyślnie Nextflow zapisuje je w $NXF_HOME/assets.
Uwaga
Pełna ścieżka może się różnić w Twoim systemie, jeśli nie używasz naszego środowiska szkoleniowego.
Nextflow celowo trzyma pobrany kod źródłowy 'z dala od drogi' w oparciu o zasadę, że te pipeline'y powinny być używane bardziej jak biblioteki niż kod, z którym bezpośrednio współdziałasz.
1.2.4. Utwórz dowiązanie symboliczne, aby łatwo uzyskać dostęp do kodu źródłowego¶
Nie będziemy szczegółowo analizować kodu, ale rzućmy na niego okiem, aby zorientować się w ogólnej organizacji.
Aby ułatwić przeglądanie kodu źródłowego pipeline'a, utwórz dowiązanie symboliczne do katalogu assets:
To tworzy skrót, który pozwala eksplorować kod za pomocą tree -L 2 pipelines lub otwierać pliki bezpośrednio.
1.2.5. Przegląd organizacji kodu¶
Możesz użyć tree lub eksploratora plików, aby znaleźć i otworzyć katalog nf-core/demo.
Zawartość katalogu
pipelines/nf-core/demo
├── assets
├── CHANGELOG.md
├── CITATIONS.md
├── CODE_OF_CONDUCT.md
├── conf
├── docs
├── LICENSE
├── main.nf
├── modules
├── modules.json
├── nextflow.config
├── nextflow_schema.json
├── nf-test.config
├── README.md
├── ro-crate-metadata.json
├── subworkflows
├── tests
├── tower.yml
└── workflows
Jak widać, dzieje się tam dużo, ale większości z tego nie musisz się martwić.
Krótko zauważmy, że na najwyższym poziomie możesz znaleźć plik README z informacjami podsumowującymi, a także pliki akcesoriów, które podsumowują informacje o projekcie, takie jak licencjonowanie, wytyczne dotyczące wkładu, cytowania i kodeks postępowania.
Szczegółowa dokumentacja pipeline'a znajduje się w katalogu docs.
Cała ta zawartość jest używana do programowego generowania stron internetowych na stronie nf-core, więc są one zawsze aktualne z kodem.
Co do reszty, możemy wyróżnić trzy funkcjonalne grupy plików kodu:
- Komponenty kodu pipeline'a (
main.nf,workflows,subworkflows,modules) - Konfiguracja pipeline'a
- Parametry pipeline'a / wejścia i walidacja
Nie będziemy omawiać komponentów kodu pipeline'a w tej części kursu, ale dotkniemy elementów konfiguracji i walidacji, które mogą być istotne dla Ciebie jako użytkownika końcowego pipeline'ów nf-core.
Wskazówka
Możesz również przeglądać kod źródłowy dowolnego pipeline'a nf-core na GitHub, np. github.com/nf-core/demo. Każdy pipeline nf-core ma taki sam układ katalogów, więc gdy już znasz tę strukturę, możesz znaleźć pliki konfiguracyjne, moduły i workflow'y dla dowolnego pipeline'a w ten sam sposób.
Ale na razie przejdźmy do uruchomienia pipeline'a!
Podsumowanie¶
Wiesz, jak znaleźć pipeline za pośrednictwem strony nf-core i pobrać lokalną kopię kodu źródłowego.
Co dalej?¶
Dowiedz się, jak wypróbować pipeline nf-core przy minimalnym wysiłku.
2. Wypróbuj pipeline z jego profilem testowym¶
Wygodnie, każdy pipeline nf-core jest dostarczany z profilem testowym. Jest to minimalny zestaw ustawień konfiguracyjnych dla pipeline'a do uruchomienia z użyciem małego zestawu danych testowych hostowanego w repozytorium nf-core/test-datasets. To świetny sposób, aby szybko wypróbować pipeline na małą skalę.
Uwaga
System profili konfiguracyjnych Nextflow pozwala łatwo przełączać się między różnymi silnikami kontenerów lub środowiskami wykonawczymi. Aby uzyskać więcej szczegółów, zobacz Hello Nextflow Część 6: Konfiguracja.
2.1. Zbadaj profil testowy¶
Dobrą praktyką jest sprawdzenie, co określa profil testowy pipeline'a przed jego uruchomieniem.
Profil test dla nf-core/demo znajduje się w pliku konfiguracyjnym conf/test.config.
Możesz go znaleźć lokalnie w kodzie źródłowym pipeline'a pobranym przez nextflow pull:
Oto zawartość tego pliku:
Od razu zauważysz, że blok komentarza na górze zawiera przykład użycia pokazujący, jak uruchomić pipeline z tym profilem testowym.
| conf/test.config | |
|---|---|
Jedyne rzeczy, które musimy dostarczyć, to to, co jest pokazane między nawiasami kątowymi w przykładowym poleceniu: <docker/singularity> i <OUTDIR>.
Przypominając, <docker/singularity> odnosi się do wyboru systemu kontenerów. Wszystkie pipeline'y nf-core są zaprojektowane tak, aby były użyteczne z kontenerami (Docker, Singularity, itp.) w celu zapewnienia powtarzalności i eliminacji problemów z instalacją oprogramowania.
Więc będziemy musieli określić, czy chcemy użyć Docker czy Singularity do testowania pipeline'a.
Część --outdir <OUTDIR> odnosi się do katalogu, w którym Nextflow zapisze wyjścia pipeline'a.
Musimy podać dla niego nazwę, którą możemy po prostu wymyślić.
Jeśli nie istnieje już, Nextflow utworzy go dla nas w czasie wykonywania.
Przechodząc do sekcji po bloku komentarza, profil testowy pokazuje nam, co zostało wstępnie skonfigurowane do testowania: przede wszystkim parametr input jest już ustawiony tak, aby wskazywał na zestaw danych testowych, więc nie musimy dostarczać własnych danych.
Jeśli podążysz za linkiem do wstępnie skonfigurowanego wejścia, zobaczysz, że jest to plik csv zawierający identyfikatory próbek i ścieżki plików dla kilku próbek eksperymentalnych.
sample,fastq_1,fastq_2
SAMPLE1_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R2.fastq.gz
SAMPLE2_PE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R2.fastq.gz
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample1_R1.fastq.gz,
SAMPLE3_SE,https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/illumina/amplicon/sample2_R1.fastq.gz,
Nazywa się to samplesheet i jest to najczęstsza forma wejścia do pipeline'ów nf-core.
Uwaga
Nie martw się, jeśli nie jesteś zaznajomiony z formatami i typami danych, nie jest to ważne dla tego, co następuje.
Więc potwierdza to, że mamy wszystko, czego potrzebujemy, aby wypróbować pipeline.
2.2. Uruchom pipeline¶
Zdecydujmy się użyć Docker dla systemu kontenerów i demo-results jako katalogu wyjściowego, i jesteśmy gotowi do uruchomienia polecenia testowego:
Wyjście polecenia
N E X T F L O W ~ version 25.10.4
Launching `https://github.com/nf-core/demo` [magical_pauling] DSL2 - revision: 45904cb9d1 [master]
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.1.0
------------------------------------------------------
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : demo-results
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Generic options
trace_report_suffix : 2025-11-21_04-57-41
Core Nextflow options
revision : master
runName : magical_pauling
containerEngine : docker
launchDir : /workspaces/training/hello-nf-core
workDir : /workspaces/training/hello-nf-core/work
projectDir : /workspaces/.nextflow/assets/nf-core/demo
userName : root
profile : docker,test
configFiles : /workspaces/.nextflow/assets/nf-core/demo/nextflow.config
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
executor > local (7)
[ff/a6976b] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) | 3 of 3 ✔
[39/731ab7] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) | 3 of 3 ✔
[7c/78d96e] NFCORE_DEMO:DEMO:MULTIQC | 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
Jeśli Twoje wyjście pasuje do tego, gratulacje! Właśnie uruchomiłeś Swój pierwszy pipeline nf-core.
Zauważysz, że jest znacznie więcej wyjścia konsoli niż podczas uruchamiania podstawowego pipeline'a Nextflow. Jest nagłówek, który zawiera podsumowanie wersji pipeline'a, wejść i wyjść oraz kilku elementów konfiguracji.
Uwaga
Twoje wyjście pokaże różne znaczniki czasu, nazwy wykonań i ścieżki plików, ale ogólna struktura i wykonanie procesów powinny być podobne.
Zwróć uwagę na linię blisko początku wyjścia:
Mówi ona, która rewizja pipeline'a została użyta.
Ponieważ nie określiliśmy wersji, Nextflow użył najnowszego commitu na gałęzi master.
Aby zapewnić powtarzalność uruchomień, powinieneś przypiąć konkretne wydanie za pomocą flagi -r:
Gwarantuje to, że za każdym razem używany jest ten sam kod pipeline'a, niezależnie od nowych commitów czy wydań.
W tym szkoleniu pomijamy -r dla uproszczenia, ale w środowisku produkcyjnym zawsze powinieneś go podawać.
Przechodząc do wyjścia wykonania, spójrzmy na linie, które mówią nam, jakie procesy zostały uruchomione:
executor > local (7)
[ff/a6976b] NFCORE_DEMO:DEMO:FASTQC (SAMPLE3_SE) | 3 of 3 ✔
[39/731ab7] NFCORE_DEMO:DEMO:SEQTK_TRIM (SAMPLE3_SE) | 3 of 3 ✔
[7c/78d96e] NFCORE_DEMO:DEMO:MULTIQC | 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
To mówi nam, że zostały uruchomione trzy procesy, odpowiadające trzem narzędziom pokazanym na stronie dokumentacji pipeline'a na stronie nf-core: FASTQC, SEQTK_TRIM i MULTIQC.
Pełne nazwy procesów, jak pokazano tutaj, takie jak NFCORE_DEMO:DEMO:MULTIQC, są dłuższe niż to, co mogłeś zobaczyć we wstępnym materiale Hello Nextflow.
Zawierają one nazwy ich workflow'ów nadrzędnych i odzwierciedlają modularność kodu pipeline'a.
Zajmiemy się tym bardziej szczegółowo w Części 2 tego kursu.
2.3. Zbadaj wyjścia pipeline'a¶
Na koniec spójrzmy na katalog demo-results wyprodukowany przez pipeline.
Zawartość katalogu
demo-results
├── fastqc
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── fq
│ ├── SAMPLE1_PE
│ ├── SAMPLE2_PE
│ └── SAMPLE3_SE
├── multiqc
│ ├── multiqc_data
│ ├── multiqc_plots
│ └── multiqc_report.html
└── pipeline_info
├── execution_report_2025-11-21_04-57-41.html
├── execution_timeline_2025-11-21_04-57-41.html
├── execution_trace_2025-11-21_04-57-41.txt
├── nf_core_demo_software_mqc_versions.yml
├── params_2025-11-21_04-57-46.json
└── pipeline_dag_2025-11-21_04-57-41.html
To może wydawać się dużo.
Aby dowiedzieć się więcej o wyjściach pipeline'a nf-core/demo, sprawdź jego stronę dokumentacji.
Na tym etapie ważne jest zaobserwowanie, że wyniki są zorganizowane według modułu, a dodatkowo istnieje katalog o nazwie pipeline_info zawierający różne raporty z datami dotyczące wykonania pipeline'a.
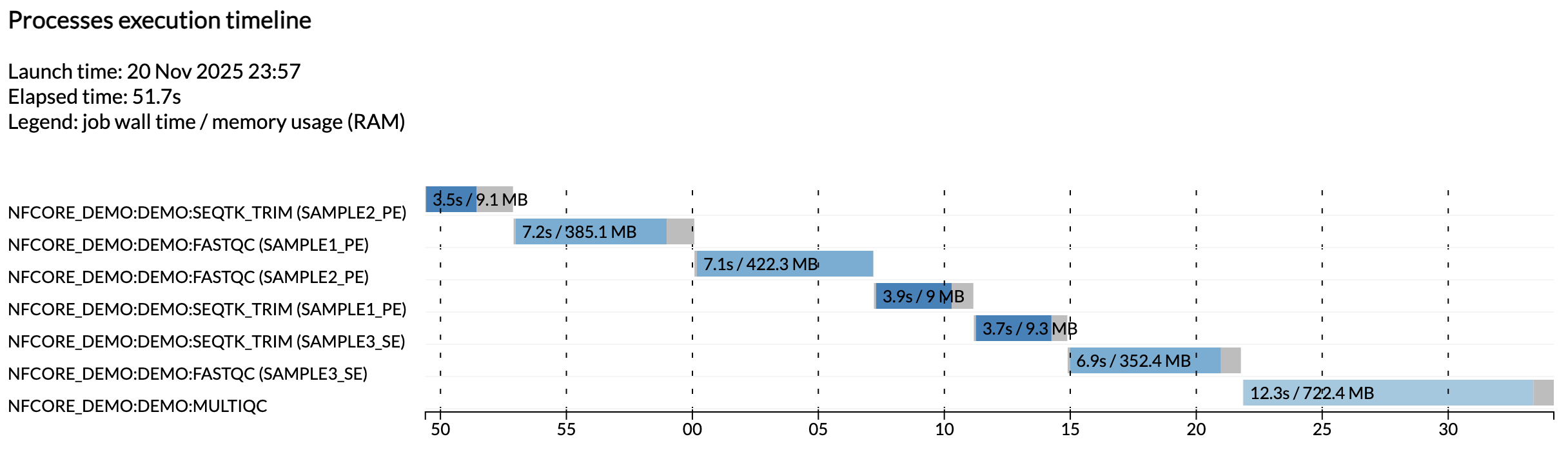
Na przykład plik execution_timeline_* pokazuje, jakie procesy zostały uruchomione, w jakiej kolejności i jak długo trwało ich uruchomienie:
Uwaga
Tutaj zadania nie zostały uruchomione równolegle, ponieważ działamy na minimalistycznej maszynie w Github Codespaces. Aby zobaczyć ich równoległe uruchomienie, spróbuj zwiększyć alokację CPU Swojego codespace i limity zasobów w konfiguracji testowej.
Te raporty są generowane automatycznie dla wszystkich pipeline'ów nf-core.
Podsumowanie¶
Wiesz, jak uruchomić pipeline nf-core używając jego wbudowanego profilu testowego i gdzie znaleźć jego wyjścia.
Co dalej?¶
Dowiedz się, jak skonfigurować pipeline, aby dostosować jego wykonanie.
3. Skonfiguruj wykonanie pipeline'a¶
Jak wyjaśniono w Hello Config, chcemy móc zmieniać dane, na których pipeline będzie działał, oraz sposób jego uruchomienia bez modyfikowania samego kodu pipeline'a. W tym celu Nextflow obsługuje wiele sposobów kontrolowania konfiguracji pipeline'a, co może być nieco przytłaczające.
Projekt nf-core określa konwencje organizowania elementów konfiguracji, rozróżniając dwa rodzaje konfiguracji na najwyższym poziomie: parametry pipeline'a i konfiguracja w ścisłym sensie.
- Parametry pipeline'a (ustawiane przez system
params) obejmują zazwyczaj takie rzeczy jak pliki wejściowe, flagi zachowania narzędzi i parametry analizy. - Konfiguracja w ścisłym sensie odnosi się do logistyki uruchamiania pipeline'a, tj. executor, alokacje zasobów obliczeniowych i tak dalej.
Zacznijmy od omówienia parametrów pipeline'a, a następnie przyjrzymy się konfiguracji w ścisłym sensie.
3.1. Parametry pipeline'a¶
Dla wszystkich pipeline'ów nf-core możesz uzyskać pełną listę parametrów bezpośrednio z wiersza poleceń, używając flagi --help, która sama w sobie jest parametrem pipeline'a.
3.1.1. Pobierz listę parametrów za pomocą --help¶
Uruchom polecenie pomocy dla pipeline'a demo:
Wyjście polecenia
N E X T F L O W ~ version 25.10.4
Launching `https://github.com/nf-core/demo` [run_name] DSL2 - revision: 45904cb9d1 [master]
----------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo 1.1.0
----------------------------------------------------
Typical pipeline command:
nextflow run nf-core/demo -profile <docker/singularity/.../institute> --input samplesheet.csv --outdir <OUTDIR>
Input/output options
--input [string] Path to a metadata file containing information about the samples in the experiment.
--outdir [string] The output directory where the results will be saved. You have to use absolute paths to storage on Cloud infrastructure.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header, used for filename if not otherwise specified.
Reference genome options
--genome [string] Name of iGenomes reference.
--fasta [string] Path to FASTA genome file.
Process skipping options
--skip_trim [boolean] Skip trimming fastq files with seqtk
Generic options
--multiqc_methods_description [string] Custom MultiQC yaml file containing HTML including a methods description.
--help [boolean, string] Display the help message.
--help_full [boolean] Display the full detailed help message.
--show_hidden [boolean] Display hidden parameters in the help message (only works when --help or --help_full are provided).
!! Hiding 20 param(s), use the `--show_hidden` parameter to show them !!
----------------------------------------------------
* The pipeline
https://doi.org/10.5281/zenodo.12192442
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
Jak widać, wyjście grupuje parametry w kategorie (opcje wejścia/wyjścia, opcje genomu referencyjnego itp.) z typami i opisami dla każdego z nich.
Ta kategoryzacja jest określana przez plik schematu, który jest omówiony dalej poniżej.
W zwykłych pipeline'ach Nextflow --help działa tylko wtedy, gdy deweloper zaimplementował to ręcznie.
Wskazówka
Użyj --help --show_hidden, aby zobaczyć dodatkowe parametry, które są domyślnie ukryte, takie jak --publish_dir_mode czy --monochrome_logs.
3.1.2. Ustaw wartości parametrów¶
Jak omówiono w Hello Config, możesz ustawiać wartości parametrów w wierszu poleceń za pomocą --nazwa_parametru lub zebrać zestaw parametrów w pliku YAML i przekazać go za pomocą -params-file.
Oba podejścia działają tak samo z pipeline'ami nf-core.
Na przykład, aby pominąć krok przycinania:
Wyjście polecenia
Proces SEQTK_TRIM nie pojawia się już w wyjściu.
Info
Choć technicznie możliwe jest ustawianie parametrów pipeline'a w niestandardowym pliku konfiguracyjnym przekazywanym za pomocą -c, może to nie nadpisywać wartości domyślnych już ustawionych w pliku nextflow.config pipeline'a, w zależności od reguł pierwszeństwa konfiguracji Nextflow.
Użycie --nazwa_parametru w wierszu poleceń lub -params-file jest bardziej niezawodne, ponieważ te zawsze mają pierwszeństwo.
Jako zasada kciuka: jeśli coś pojawia się w wyjściu --help, ustaw to przez wiersz poleceń lub plik parametrów, a nie plik konfiguracyjny.
3.1.3. Walidacja parametrów¶
Ciekawostka: polecenie --help działa dla wszystkich pipeline'ów nf-core, ponieważ projekt nf-core wymaga od deweloperów formalnego zdefiniowania wszystkich parametrów pipeline'a w pliku schematu JSON (nextflow_schema.json).
Ten schemat rejestruje typ, opis, wartość domyślną i grupowanie każdego parametru.
Oprócz zasilania wyjścia --help, plik schematu umożliwia również automatyczną walidację podczas uruchamiania.
Oznacza to, że Nextflow może sprawdzić, czy każdy przekazany parametr istnieje i ma odpowiednią wartość (odpowiedniego typu, w dozwolonym zakresie wartości itp.).
Omawiamy to bardziej szczegółowo w Części 5: Walidacja wejścia, ale możesz już zobaczyć to w działaniu, podając pipeline'owi demo nieprawidłowe dane wejściowe.
3.1.3.1. Nierozpoznane parametry¶
Spróbuj przekazać parametr, który nie istnieje:
Wyjście konsoli zawiera ostrzeżenie:
Pipeline nadal działa, ale ostrzeżenie natychmiast informuje Cię, że --foobar nie jest rozpoznanym parametrem.
Wyłapuje to literówki, takie jak --outDir zamiast --outdir, zanim zmarnujesz czas obliczeniowy zastanawiając się, dlaczego wyjście trafiło w złe miejsce.
3.1.3.2. Nieprawidłowe wartości parametrów¶
Walidacja sprawdza również wartości parametrów.
Parametr --skip_trim jest flagą boolean, więc przekazanie wartości string powoduje natychmiastowe niepowodzenie pipeline'a:
ERROR ~ Validation of pipeline parameters failed!
The following invalid input values have been detected:
* --skip_trim (yes): Value is [string] but should be [boolean]
Pipeline zatrzymuje się przed uruchomieniem jakichkolwiek procesów, oszczędzając Cię przed nieudanym lub nieprawidłowym wykonaniem.
Parametry boolean powinny być przekazywane jako flagi (--skip_trim) bez wartości lub ustawiane na true/false w pliku parametrów.
3.1.4. Walidacja wejścia¶
Ta sama logika walidacji może być również używana do sprawdzania poprawności plików wejściowych. Na przykład, jeśli pipeline oczekuje samplesheet jako głównego wejścia danych (co ma miejsce w przypadku wielu, jeśli nie większości pipeline'ów nf-core), deweloper może dostarczyć schemat wejścia (odrębny od schematu parametrów) opisujący, jak powinien być zorganizowany plik wejściowy.
Następnie, w czasie wykonywania, Nextflow może sprawdzić, czy dostarczony plik wejściowy jest prawidłowy.
Omawiamy to również bardziej szczegółowo w Części 5: Walidacja wejścia, ale możesz już zobaczyć to w działaniu, podając pipeline'owi demo nieprawidłowy samplesheet.
Pipeline nf-core/demo oczekuje pliku CSV z kolumnami sample, fastq_1 i fastq_2.
Jest to zdefiniowane w pliku schematu (assets/schema_input.json), który określa oczekiwaną strukturę, typy kolumn i ograniczenia.
assets/schema_input.json
{
"$schema": "https://json-schema.org/draft/2020-12/schema",
"$id": "https://raw.githubusercontent.com/nf-core/demo/master/assets/schema_input.json",
"title": "nf-core/demo pipeline - params.input schema",
"description": "Schema for the file provided with params.input",
"type": "array",
"items": {
"type": "object",
"properties": {
"sample": {
"type": "string",
"pattern": "^\\S+$",
"errorMessage": "Sample name must be provided and cannot contain spaces",
"meta": ["id"]
},
"fastq_1": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 1 must be provided, cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
},
"fastq_2": {
"type": "string",
"format": "file-path",
"exists": true,
"pattern": "^([\\S\\s]*\\/)?[^\\s\\/]+\\.f(ast)?q\\.gz$",
"errorMessage": "FastQ file for reads 2 cannot contain spaces and must have extension '.fq.gz' or '.fastq.gz'"
}
},
"required": ["sample", "fastq_1"]
}
}
Schemat określa, że sample i fastq_1 są wymagane, podczas gdy fastq_2 jest opcjonalne (obsługując zarówno dane paired-end, jak i single-end).
Ścieżki plików są walidowane pod kątem istnienia i wzorca rozszerzenia.
3.1.4.1. Utwórz nieprawidłowy samplesheet¶
Utwórz samplesheet z brakującą kolumną i nieistniejącą ścieżką pliku:
Ten samplesheet nie ma wymaganej kolumny fastq_1 i zawiera nieistniejącą ścieżkę pliku w fastq_2.
Oba problemy spowodują błędy walidacji w następnym kroku.
3.1.4.2. Uruchom pipeline demo z nieprawidłowym samplesheet'em¶
Uruchom pipeline demo używając malformed_samplesheet.csv jako wejścia.
nextflow run nf-core/demo -profile docker,test --outdir demo-results --input malformed_samplesheet.csv
ERROR ~ Validation of pipeline parameters failed!
The following invalid input values have been detected:
* --input (malformed_samplesheet.csv): Validation of file failed:
-> Entry 1: Error for field 'fastq_2' (/not/a/real/file.fastq.gz): the file or directory
'/not/a/real/file.fastq.gz' does not exist (FastQ file for reads 2 cannot contain spaces
and must have extension '.fq.gz' or '.fastq.gz')
-> Entry 1: Missing required field(s): fastq_1
Jak widać, pipeline natychmiast kończy się niepowodzeniem i zgłasza wszystkie błędy walidacji naraz. nf-schema nie zatrzymuje się na pierwszym błędzie — zbiera każdy problem i wyświetla je razem, dzięki czemu możesz naprawić wszystko za jednym razem, zamiast odkrywać problemy jeden po drugim.
Każdy błąd identyfikuje dokładny wpis i pole, które spowodowało problem, więc możesz naprawić swój samplesheet, a następnie ponownie uruchomić pipeline z pewnością, że nie zakończy się niepowodzeniem w późniejszym momencie, gdy Nextflow faktycznie spróbuje uzyskać dostęp do ścieżki pliku.
Dla deweloperów wszystko to jest omówione bardziej szczegółowo w Części 5 tego kursu.
3.2. Konfiguracja¶
Konfiguracja w ścisłym sensie kontroluje sposób uruchamiania pipeline'a: alokację zasobów, argumenty specyficzne dla narzędzi, miejsce wykonywania zadań i używany system pakowania oprogramowania.
Pipeline'y nf-core zawierają domyślną konfigurację w nextflow.config i katalogu conf/.
Przed nadpisaniem czegokolwiek warto wiedzieć, gdzie znajdują się wartości domyślne.
Widziałeś już w sekcji 2.1, że kod źródłowy pipeline'a znajduje się w $NXF_HOME/assets.
Wylistuj pliki konfiguracyjne, aby zobaczyć, co jest dostępne:
Najważniejsze pliki konfiguracyjne to:
conf/base.config: Definiuje etykiety zasobów (process_low,process_medium,process_high), które przypisują procesory, pamięć i czas do procesów. Gdy widzisz proces używający więcej zasobów niż oczekiwano, stąd pochodzą te wartości domyślne.conf/modules.config: Ustawia argumenty narzędzi dla poszczególnych procesów (ext.args) i ustawienia publikowania wyjść (publishDir). Otwórz ten plik, aby zobaczyć, jakie argumenty każde narzędzie otrzymuje domyślnie.conf/test.config: Profil testowy użyty w sekcji 2.1, który ogranicza zasoby za pomocąresourceLimitsi ustawia testowy samplesheet. Aktywowany za pomocą-profile test. Istnieje równieżconf/test_full.configdo uruchamiania z pełnowymiarowym zestawem danych testowych, przydatny do benchmarkingu.
Centralny plik nextflow.config ładuje wszystkie powyższe i ustawia odpowiednie wartości domyślne dla wszystkiego.
Jeśli chcesz zmodyfikować którekolwiek z ustawień określonych w tych plikach, nie modyfikuj żadnego z nich bezpośrednio.
Zamiast tego utwórz własny plik konfiguracyjny i przekaż go za pomocą -c.
Wartości, które określisz, nadpiszą wartości domyślne ustawione w tych innych plikach.
Przejdźmy przez kilka ćwiczeń, aby zrobić to w praktyce.
3.2.1. Zmień alokację zasobów dla procesu¶
Pipeline demo przypisuje zasoby za pomocą etykiet zdefiniowanych w base.config.
Na przykład FASTQC używa etykiety process_medium, która przydziela 6 procesorów i 36 GB pamięci.
Profil testowy ogranicza zasoby za pomocą resourceLimits, ale możesz również nadpisać zasoby dla konkretnych procesów.
Utwórz plik o nazwie custom.config:
Uruchom pipeline z Twoją niestandardową konfiguracją:
Wyjście polecenia
Flaga -c dodaje Twoją konfigurację na wierzch wbudowanej konfiguracji pipeline'a.
3.2.2. Ustaw wartości argumentów narzędzi za pomocą ext.args¶
Wiele narzędzi wiersza poleceń ma argumenty, które nie są wymagane i dlatego nie są konfigurowane jako parametry pipeline'a, chyba że są bardzo często używane.
Dla tych argumentów narzędzi moduły nf-core używają konwencji Nextflow o nazwie ext.args, aby przekazywać argumenty do bazowego narzędzia przez plik konfiguracyjny.
Na przykład dodajmy argument przycinania do modułu SEQTK_TRIM za pomocą ext.args.
3.2.2.1. Zaktualizuj niestandardową konfigurację¶
Zaktualizuj swój plik custom.config:
| custom.config | |
|---|---|
To mówi seqtk trimfq, aby przyciął 5 zasad od początku każdego odczytu, oprócz przycinania jakościowego.
3.2.2.2. Uruchom pipeline¶
Uruchom pipeline ponownie z tą konfiguracją, aby zobaczyć efekt:
Wyjście polecenia
Aby zweryfikować, że argument został zastosowany, znajdź hash katalogu roboczego SEQTK_TRIM z wyjścia uruchomienia (np. work/ab/cd1234...) i sprawdź plik .command.sh w jego wnętrzu:
Wyjście polecenia
Powinieneś zobaczyć -b 5 w poleceniu seqtk trimfq, co potwierdza, że Twoje nadpisanie ext.args zadziałało.
3.2.2.3. Nadpisywanie wartości domyślnych¶
Niektóre moduły mają już domyślnie ustawione ext.args.
Na przykład moduł FASTQC jest domyślnie skonfigurowany z ext.args = '--quiet' (zdefiniowanym w conf/modules.config).
| conf/modules.config | |
|---|---|
Jeśli podasz wartość dla ext.args przez niestandardowy plik konfiguracyjny, ta wartość całkowicie zastąpi wartość domyślną ustawioną dla tego procesu.
Tak więc na przykład, jeśli wartość domyślna to '--quiet' i ustawisz ext.args = '--kmers 8', flaga --quiet nie będzie już stosowana.
Aby zachować obie, ustaw ext.args = '--quiet --kmers 8'.
Oznacza to, że jesteś odpowiedzialny za sprawdzenie, jaka jest domyślna konfiguracja narzędzi, do których chcesz dostarczyć wartości argumentów za pomocą ext.args.
Podsumowanie¶
Wiesz, jak uzyskać pomoc od pipeline'a nf-core, ustawiać parametry i rozumieć, jak są walidowane, oraz dostosowywać konfigurację przez pliki konfiguracyjne.
Co dalej?¶
Zrób sobie przerwę! Gdy będziesz gotowy, przejdź do Części 2, gdzie stworzysz własny pipeline kompatybilny z nf-core od podstaw.